






































































































































































































































































































よむ、つかう、まなぶ。
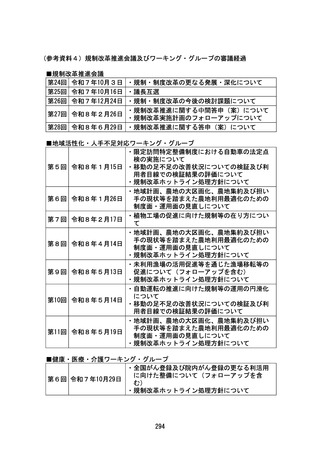
資料2 規制改革推進に関する答申(案) (54 ページ)
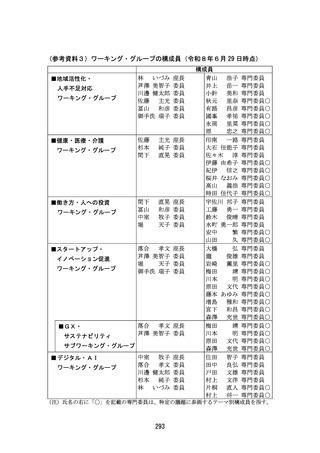
出典
| 公開元URL | https://www8.cao.go.jp/kisei-kaikaku/kisei/meeting/committee/260629/agenda.html |
| 出典情報 | 規制改革推進会議(第28回 6/29)《内閣府》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
カ
被験者保護及び研究力強化等のための倫理審査の適正化
【a:令和8年度上期結論、結論を得次第速やかに措置、
b:①令和8年措置、②令和8年度上期結論、結論を得次第速やかに措置、
③(前段)令和8年度上期結論、結論を得次第速やかに措置、
(後段)令和 11 年度措置、c:a の措置から5年間継続的に措置、
d:令和8年度措置】
<実施事項>
近年、厳しい国際競争の中で我が国の創薬力の低下が繰り返し指摘されて
いる。また、令和5年3月時点において、平成 28 年から令和2年に欧米で
承認された新医薬品のうち我が国で承認されていない医薬品が 143 品目存
在していたなど、ドラッグ・ラグ(欧米では承認されているが日本では承認
されていない医薬品が発生している事象のことをいう。以下同じ。)/ドラ
ッグ・ロス(欧米では承認されているが日本では承認されていない医薬品が
発生している事象のうち、特に日本での開発に着手されていないものをいう。
以下同じ。)の問題が指摘されている。これらの課題解決には、我が国での
国際共同治験の実施が重要であるが、令和4年度以降の我が国での国際共同
治験の実施数は、令和2年度及び令和3年度に比べて減少傾向である。我が
国の創薬力を強化するとともに、ドラッグ・ラグ/ドラッグ・ロスを解消す
るためには、我が国の創薬研究の実施環境を欧米にそん色ない魅力あるもの
とし、我が国での国際共同治験の実施を推進していく必要がある。
こうした中、令和6年6月の規制改革実施計画では、内閣府、こども家庭
庁、文部科学省、厚生労働省及び経済産業省(以下「関係府省」という。)
は、①我が国において、医薬品、医療機器等の品質、有効性及び安全性の確
保等に関する法律(昭和 35 年法律第 145 号)が適用される治験、臨床研究
法(平成 29 年法律第 16 号)が適用される臨床研究(以下「臨床研究」とい
う。)、人を対象とする生命科学・医学系研究に関する倫理指針(令和3年文
部科学省・厚生労働省・経済産業省告示第1号)等が適用される研究等(以
下「生命科学・医学研究等」といい、また、治験、臨床研究及び生命科学・
医学研究等を総称して「治験・研究」という。以下同じ。
)を行う場合には、
海外と異なり、その目的と種類によって適用される法規制が異なっているこ
となどを背景として、治験・研究の内容によって異なる対応(異なる委員会
による審査への対応を含む。)が求められることが大きな負荷となっている
ことや倫理審査委員会等の審査の質のばらつき等の一因になっているなど
の指摘があること、②我が国の治験パフォーマンスは海外に比べ低く、また、
治験環境は海外に比べコスト面での違いが大きいとの指摘や、国際共同治験
において我が国が選ばれないことがドラッグ・ラグ/ドラッグ・ロスの一因
53
被験者保護及び研究力強化等のための倫理審査の適正化
【a:令和8年度上期結論、結論を得次第速やかに措置、
b:①令和8年措置、②令和8年度上期結論、結論を得次第速やかに措置、
③(前段)令和8年度上期結論、結論を得次第速やかに措置、
(後段)令和 11 年度措置、c:a の措置から5年間継続的に措置、
d:令和8年度措置】
<実施事項>
近年、厳しい国際競争の中で我が国の創薬力の低下が繰り返し指摘されて
いる。また、令和5年3月時点において、平成 28 年から令和2年に欧米で
承認された新医薬品のうち我が国で承認されていない医薬品が 143 品目存
在していたなど、ドラッグ・ラグ(欧米では承認されているが日本では承認
されていない医薬品が発生している事象のことをいう。以下同じ。)/ドラ
ッグ・ロス(欧米では承認されているが日本では承認されていない医薬品が
発生している事象のうち、特に日本での開発に着手されていないものをいう。
以下同じ。)の問題が指摘されている。これらの課題解決には、我が国での
国際共同治験の実施が重要であるが、令和4年度以降の我が国での国際共同
治験の実施数は、令和2年度及び令和3年度に比べて減少傾向である。我が
国の創薬力を強化するとともに、ドラッグ・ラグ/ドラッグ・ロスを解消す
るためには、我が国の創薬研究の実施環境を欧米にそん色ない魅力あるもの
とし、我が国での国際共同治験の実施を推進していく必要がある。
こうした中、令和6年6月の規制改革実施計画では、内閣府、こども家庭
庁、文部科学省、厚生労働省及び経済産業省(以下「関係府省」という。)
は、①我が国において、医薬品、医療機器等の品質、有効性及び安全性の確
保等に関する法律(昭和 35 年法律第 145 号)が適用される治験、臨床研究
法(平成 29 年法律第 16 号)が適用される臨床研究(以下「臨床研究」とい
う。)、人を対象とする生命科学・医学系研究に関する倫理指針(令和3年文
部科学省・厚生労働省・経済産業省告示第1号)等が適用される研究等(以
下「生命科学・医学研究等」といい、また、治験、臨床研究及び生命科学・
医学研究等を総称して「治験・研究」という。以下同じ。
)を行う場合には、
海外と異なり、その目的と種類によって適用される法規制が異なっているこ
となどを背景として、治験・研究の内容によって異なる対応(異なる委員会
による審査への対応を含む。)が求められることが大きな負荷となっている
ことや倫理審査委員会等の審査の質のばらつき等の一因になっているなど
の指摘があること、②我が国の治験パフォーマンスは海外に比べ低く、また、
治験環境は海外に比べコスト面での違いが大きいとの指摘や、国際共同治験
において我が国が選ばれないことがドラッグ・ラグ/ドラッグ・ロスの一因
53