


































































































よむ、つかう、まなぶ。
資料5-1 Ⅳ-203 モキシフロキサシン塩酸塩[15.1MB] (21 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/shingi2/0000198856_00044.html |
| 出典情報 | 医療上の必要性の高い未承認薬・適応外薬検討会議(第66回 12/12)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
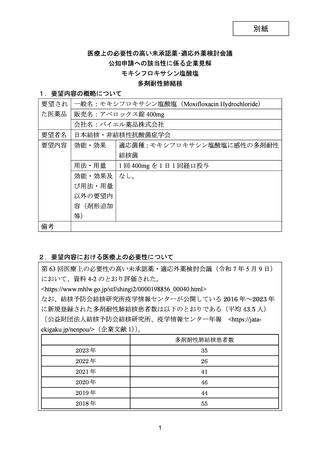
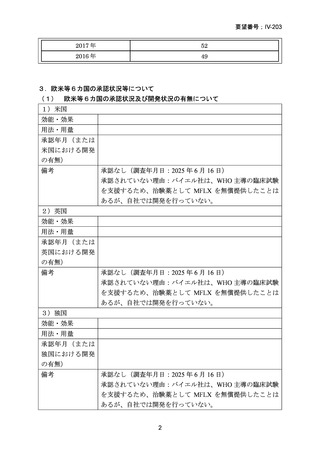
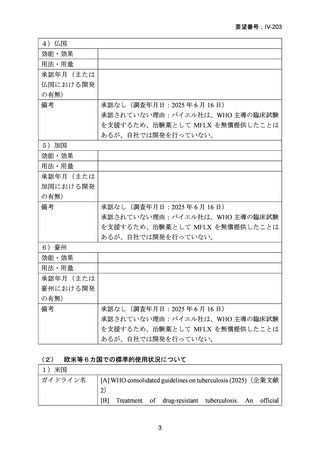
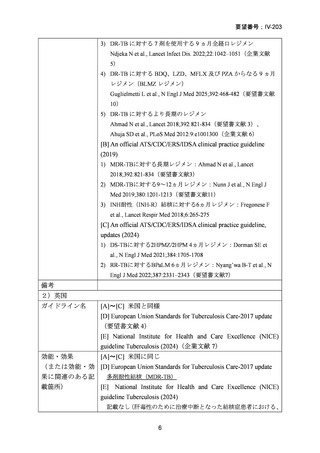
要望番号;IV-203
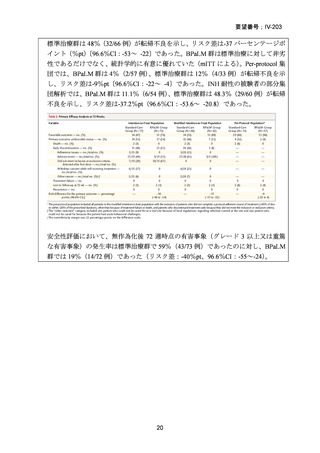
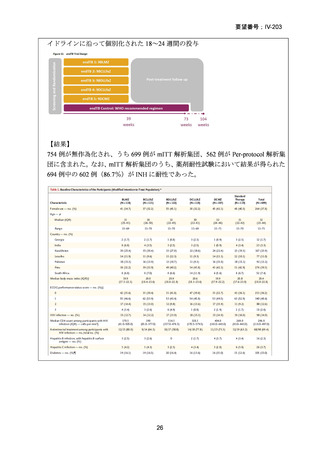
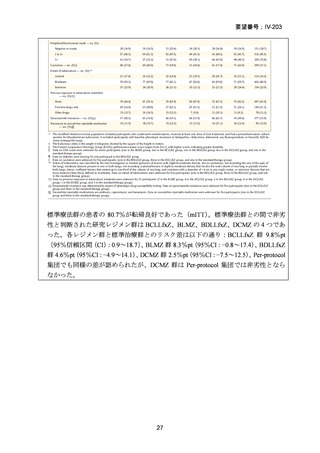
性評価として治療終了時(研究レジメン群では 24 週間、SoC 群では 36~80+週間)
における SAE 又はグレード 3 以上の AE が認められた被験者の割合が含まれてい
た。Stage 2 での目標症例数は、研究レジメン群、SoC 群ともそれぞれ 201 例(Stage
1 からの移行症例を含む)であった。
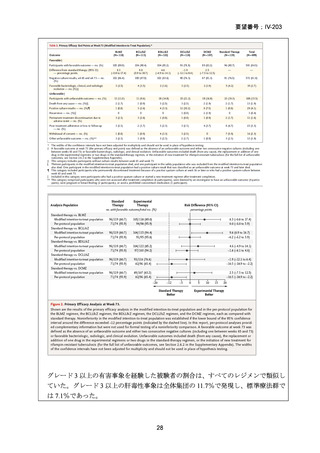
【対象患者】
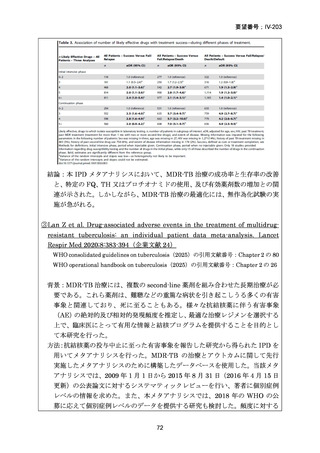
主な選択基準
15 歳以上の男女で、HIV の状態は問わない
結核菌(Mycobacterium tuberculosis)の存在が微生物学的検査(分子又は表現型)
で確認されている
分子又は表現型の薬剤感受性試験により、RFP 耐性が確認された場合
主な除外基準
治験薬に対する既知のアレルギー、過敏症、又は不耐症
妊娠中、授乳中、又は適切な避妊措置を使用することに消極的な場合
肝酵素が正常上限の 3 倍を超えている場合
QTcF > 450ms
QTc 延長のリスク因子が 1 つ以上(年齢及び性別を除く)又は TdP に対する他
の未修正のリスク因子;
心疾患の既往、失神エピソード、症候性又は無症候性の不整脈(洞性不整脈を
除く);
ベースライン検査値がグレード 4 の毒性に一致する場合
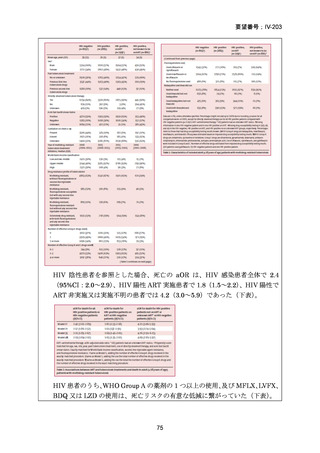
BDQ、プレトマニド、LZD 又は DLM に対する既知の耐性
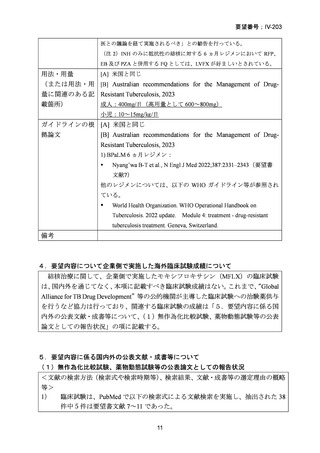
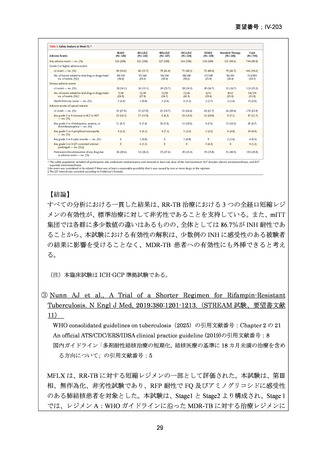
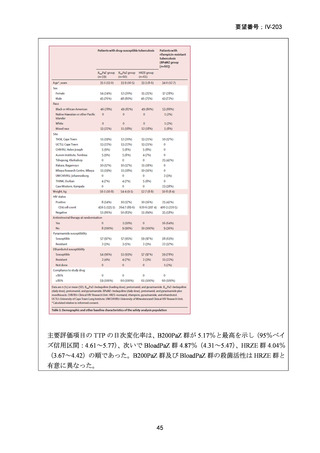
【治療群】
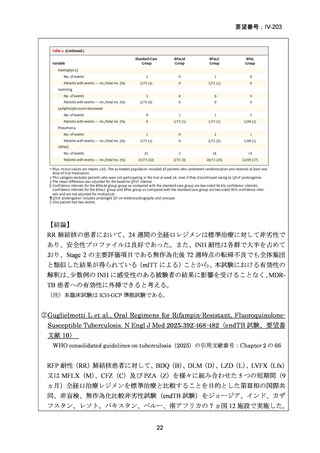
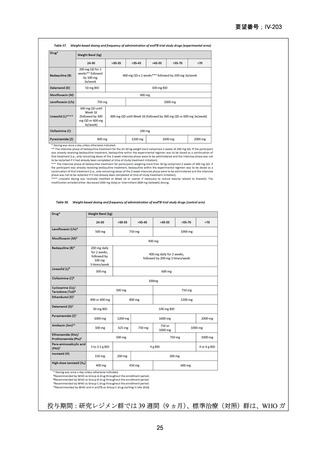
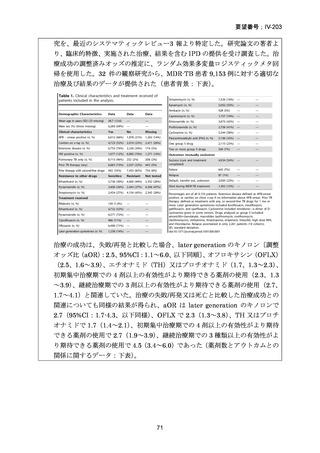
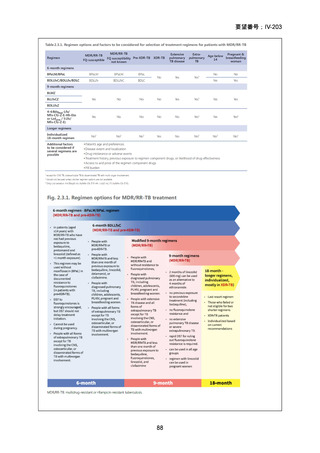
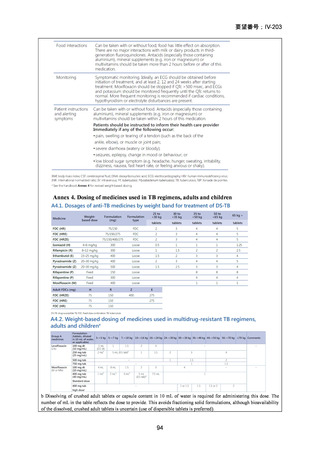
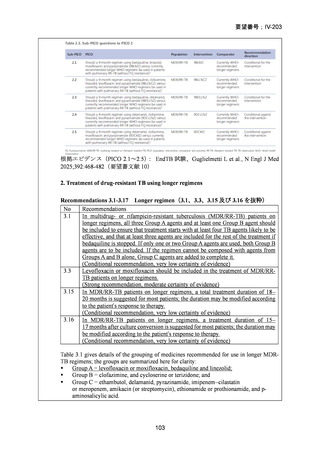
Stage 1 では、研究レジメンとして以下の 3 つ、すなわち、レジメン1:BPaLM、レ
ジメン 2:BPaLC、レジメン 3:BPaL が投与された。各薬剤の用法・用量は以下の
とおりで、すべて経口投与であった。治療期間は無作為化後 24 週間、追跡期間は 84
週までであった。
対照群は、ローカルで承認されている標準治療で、可能な限り WHO で推奨される
M/XDR-TB の治療に沿うものとした。治療期間は 36 週間以上であるが、それぞれ
の正確な期間は現行の WHO ガイドラインに基づく治療反応に依存していた。
17
性評価として治療終了時(研究レジメン群では 24 週間、SoC 群では 36~80+週間)
における SAE 又はグレード 3 以上の AE が認められた被験者の割合が含まれてい
た。Stage 2 での目標症例数は、研究レジメン群、SoC 群ともそれぞれ 201 例(Stage
1 からの移行症例を含む)であった。
【対象患者】
主な選択基準
15 歳以上の男女で、HIV の状態は問わない
結核菌(Mycobacterium tuberculosis)の存在が微生物学的検査(分子又は表現型)
で確認されている
分子又は表現型の薬剤感受性試験により、RFP 耐性が確認された場合
主な除外基準
治験薬に対する既知のアレルギー、過敏症、又は不耐症
妊娠中、授乳中、又は適切な避妊措置を使用することに消極的な場合
肝酵素が正常上限の 3 倍を超えている場合
QTcF > 450ms
QTc 延長のリスク因子が 1 つ以上(年齢及び性別を除く)又は TdP に対する他
の未修正のリスク因子;
心疾患の既往、失神エピソード、症候性又は無症候性の不整脈(洞性不整脈を
除く);
ベースライン検査値がグレード 4 の毒性に一致する場合
BDQ、プレトマニド、LZD 又は DLM に対する既知の耐性
【治療群】
Stage 1 では、研究レジメンとして以下の 3 つ、すなわち、レジメン1:BPaLM、レ
ジメン 2:BPaLC、レジメン 3:BPaL が投与された。各薬剤の用法・用量は以下の
とおりで、すべて経口投与であった。治療期間は無作為化後 24 週間、追跡期間は 84
週までであった。
対照群は、ローカルで承認されている標準治療で、可能な限り WHO で推奨される
M/XDR-TB の治療に沿うものとした。治療期間は 36 週間以上であるが、それぞれ
の正確な期間は現行の WHO ガイドラインに基づく治療反応に依存していた。
17