

































































よむ、つかう、まなぶ。
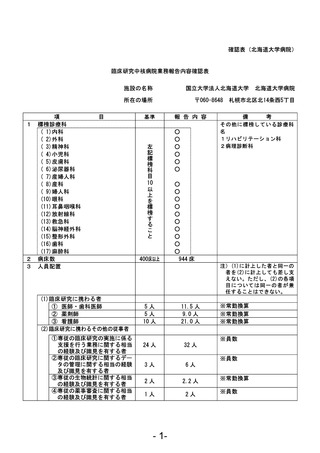
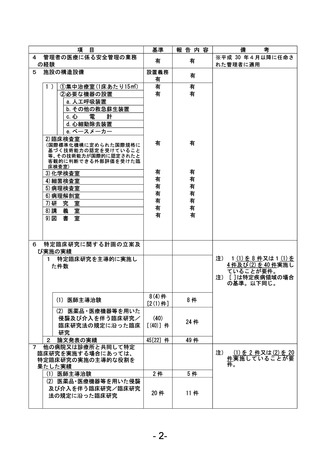
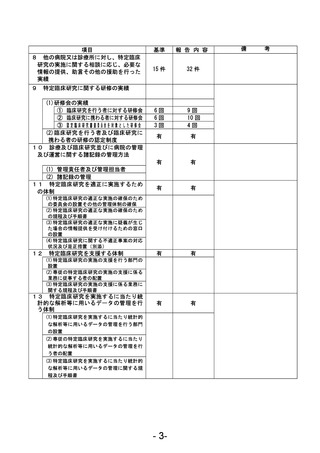
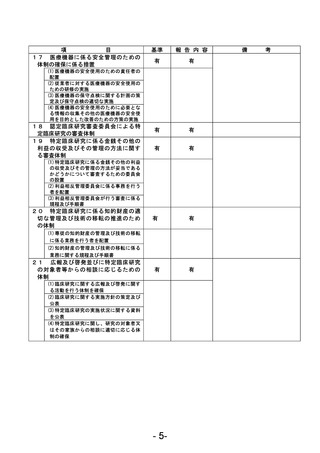
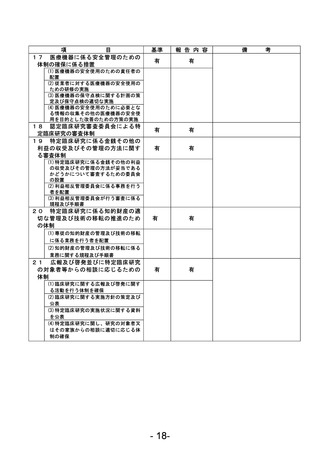
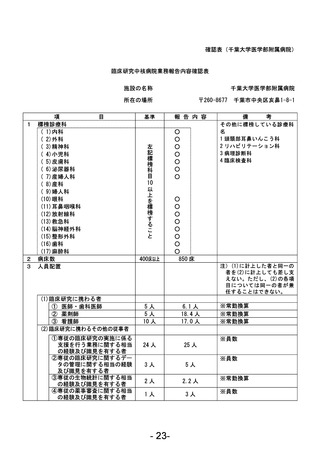
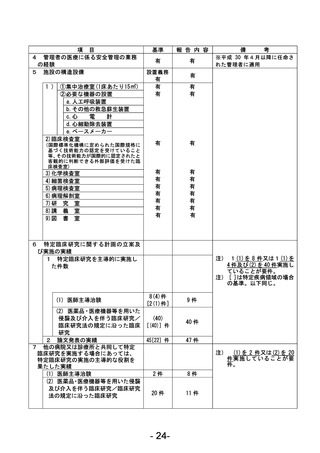
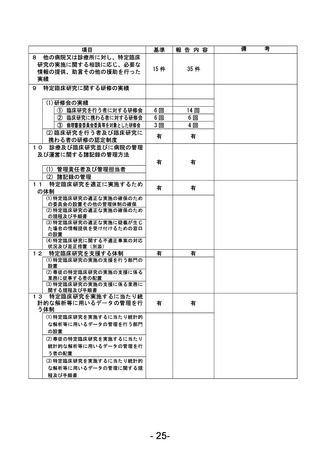
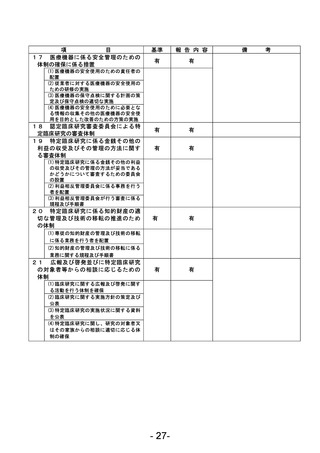
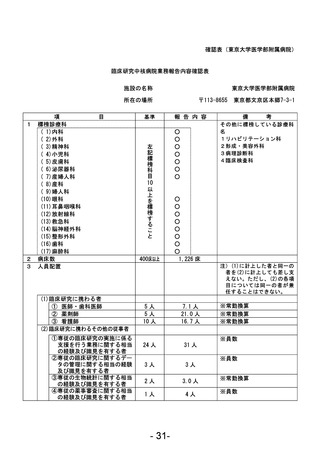
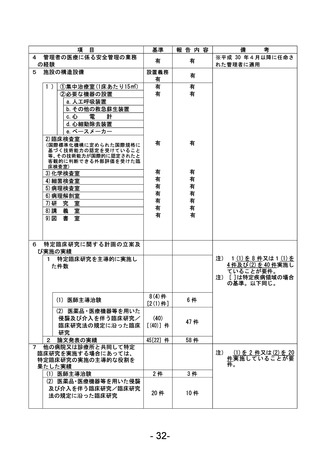
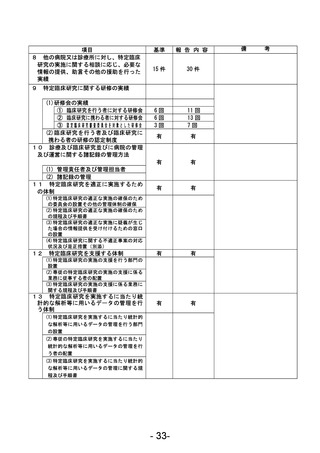
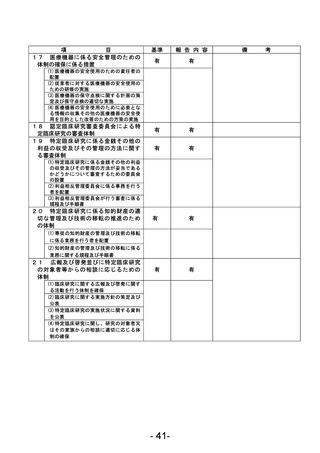
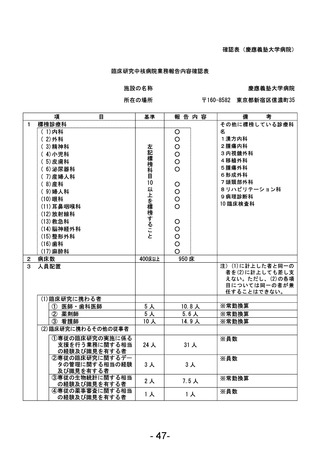
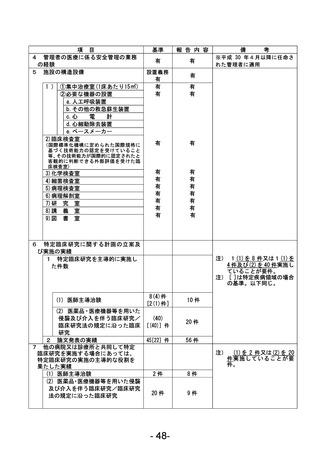
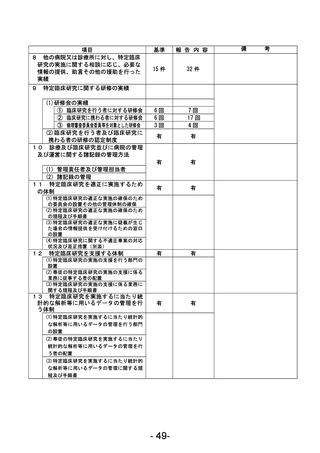
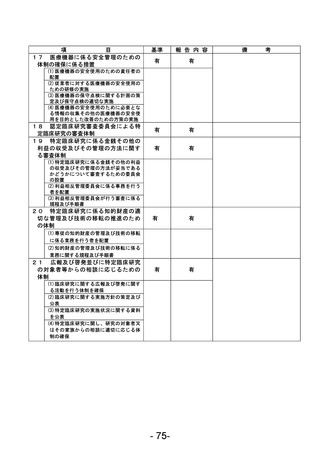
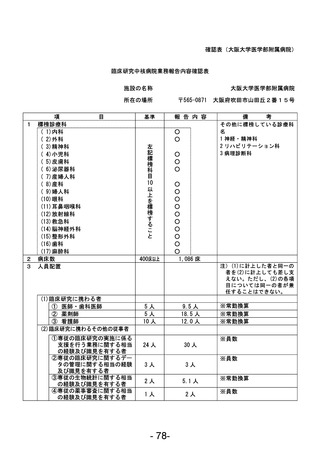
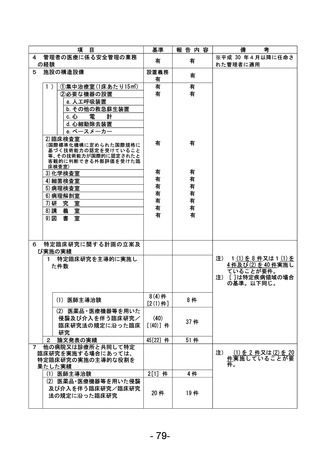
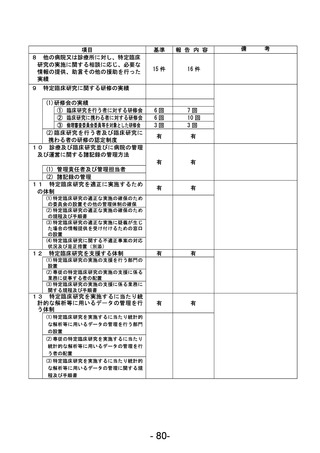
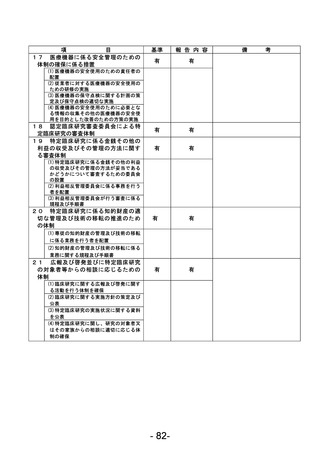
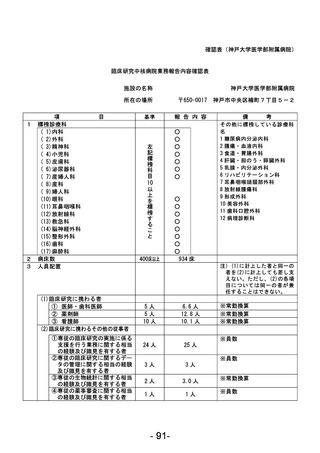
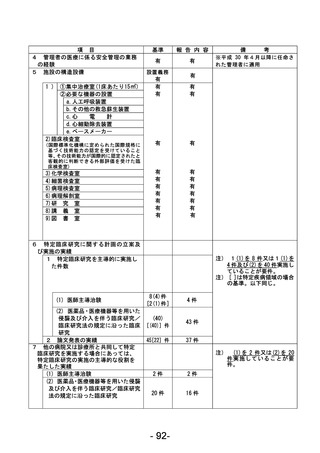
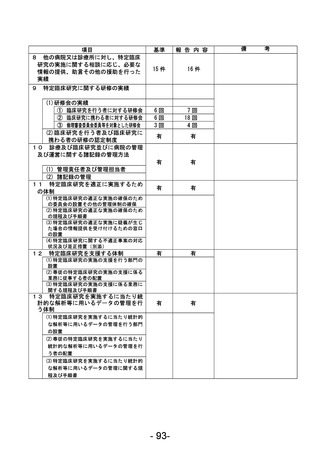
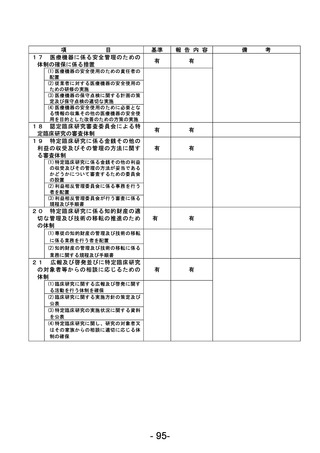
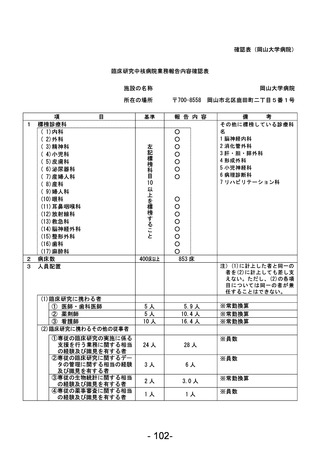
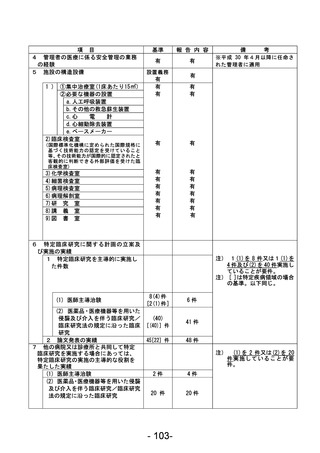
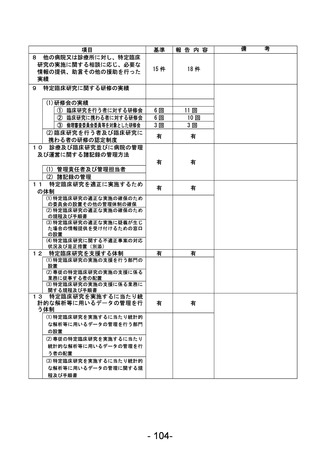
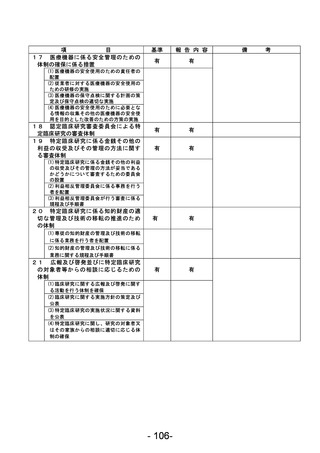
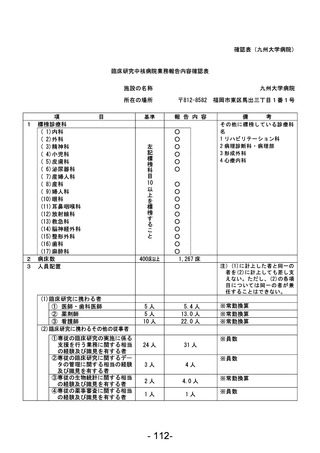
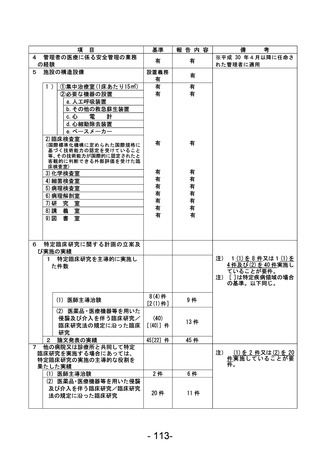
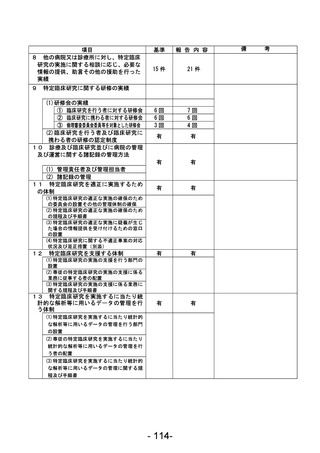
資料1 臨床研究中核病院業務報告内容確認表 (90 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_62409.html |
| 出典情報 | 厚生科学審議会 臨床研究部会(第44回 8/27)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
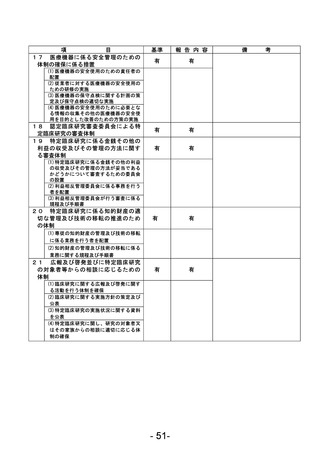
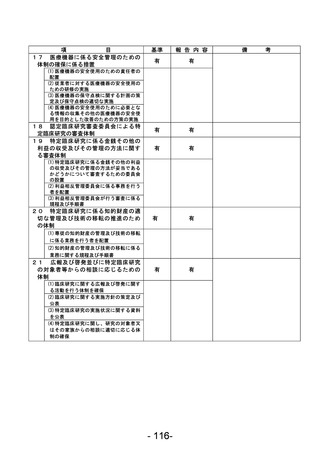
(様式第7)
登録 ID 等
****
治験・臨床研究名
****
不適正事案の概要:未承認の同意説明文書を用いて同意取得をした。
IRBからの指示、確認事項をもとに同意説明文書①(第2版 作成日:2022年7月21日)をIRB事務局へ
提出、再度IRB委員より追加の指摘事項があり同意説明文書②(第2版 作成日:2022年8月13日)を
提出、2022年8月19日に病院長より承認を得た。2023年12月8日SDV時にCRAが全症例の同意書を確認し
た際、うち1名が同意説明文書①を用いて文書取得していることが判明した。
<本件に対する責任医師見解>
同意説明文書①から同意説明文書②への改訂内容について、本被験者の年齢および本疾患の特性上、
被験者が精子凍結を行う可能性は極めて低く、本改訂内容の説明が遅延したことによる被験者の不
利益はなかったと判断する。
不適正事案に関する対応状況:
判明後、同意説明文書①から同意説明文書②の改訂内容を説明し、同意説明文書②による再同意を取
得した。
是正措置:
・2023年6月より運用開始された同意取得時チェックリストの使用を徹底する。
・以下の整合性をCRC2名でダブルチェックする。
<初回申請の場合>
病院長承認済みの治験実施計画書等変更報告書(書式6)に記載されている同意説明文書の版数、作
成日と実際に使用するために準備した同意説明文書の版数、作成日の整合性を確認する。
<変更申請の場合>
IRB開催後、同日中に配信される「IRB審査結果のお知らせ」メールを受信後、治験業務支援システム
に掲載されている同意説明文書の版数、作成日と実際に使用するために準備した同意説明文書の版
数、作成日の整合性を確認する。
なお、確認した時点で、旧版の同意説明文書は破棄する
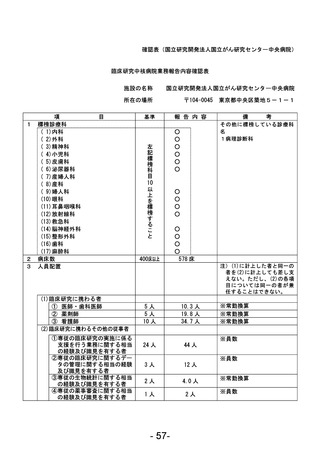
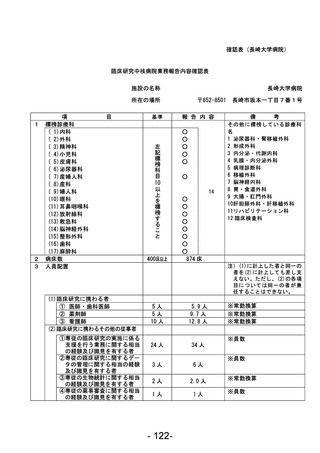
登録 ID 等
****
治験・臨床研究名
****
不適正事案の概要:
2024 年 1 月に変更申請において、研究期間 2024/3/31 から 2026/3/31、登録期間 2023/3/31 から
2025/3/31 を変更する際に、事務局確認において、2023/3/31 までの登録期間の終了に対して、登録
期間外に同意取得・登録したことが判明した。
2024 年 2 月 CRB にて登録期間外に同意取得した件について、被験者の権利保護にかかわるという点
も踏まえ、本件を重大として考えているか否かについて、研究代表医師に見解を求めたところ、重大
な不適合報告として提出する旨となった。
不適正事案に関する対応状況:
変更申請を提出。本件に参加頂いた患者の研究参加のご意思も鑑み、当該症例には「研究期間内では
あったが登録期間外の登録となった」旨をご説明の上、引き続き登録症例として経過観察を継続する
旨とした。
是正措置:
登録期間終了が研究期間終了よりも 1 年早いという認識が欠如していたこと、毎回の症例登録の際
に登録期間の確認を行っていなかったことが理由と考えられる。再発防止策としては、登録期間の認
識を改めて徹底するとともに、該当する時期に登録終了日の再確認および延長申請の必要性をリマ
インドさせるメールが届くように設定した。
CRB としては総括報告書を提出する際には、全症例の解析データとは別途、登録期間外に登録した症
例を除外した解析データも記載することもコメントとして挙げている。
(注)1
不適正事案に関する調査の概要(方法、期間、結果等)を記載すること。
- 89-
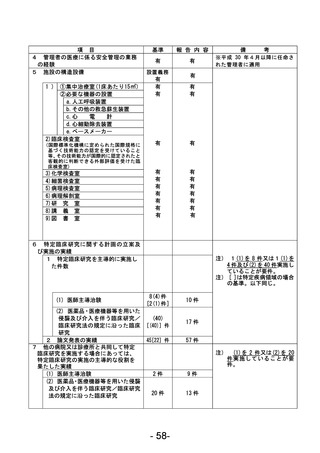
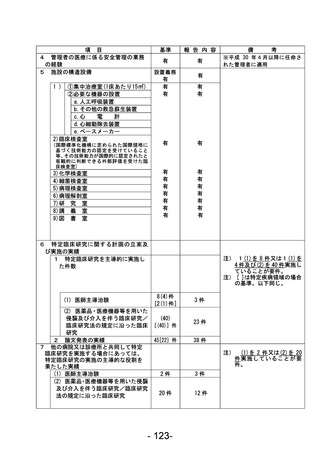
登録 ID 等
****
治験・臨床研究名
****
不適正事案の概要:未承認の同意説明文書を用いて同意取得をした。
IRBからの指示、確認事項をもとに同意説明文書①(第2版 作成日:2022年7月21日)をIRB事務局へ
提出、再度IRB委員より追加の指摘事項があり同意説明文書②(第2版 作成日:2022年8月13日)を
提出、2022年8月19日に病院長より承認を得た。2023年12月8日SDV時にCRAが全症例の同意書を確認し
た際、うち1名が同意説明文書①を用いて文書取得していることが判明した。
<本件に対する責任医師見解>
同意説明文書①から同意説明文書②への改訂内容について、本被験者の年齢および本疾患の特性上、
被験者が精子凍結を行う可能性は極めて低く、本改訂内容の説明が遅延したことによる被験者の不
利益はなかったと判断する。
不適正事案に関する対応状況:
判明後、同意説明文書①から同意説明文書②の改訂内容を説明し、同意説明文書②による再同意を取
得した。
是正措置:
・2023年6月より運用開始された同意取得時チェックリストの使用を徹底する。
・以下の整合性をCRC2名でダブルチェックする。
<初回申請の場合>
病院長承認済みの治験実施計画書等変更報告書(書式6)に記載されている同意説明文書の版数、作
成日と実際に使用するために準備した同意説明文書の版数、作成日の整合性を確認する。
<変更申請の場合>
IRB開催後、同日中に配信される「IRB審査結果のお知らせ」メールを受信後、治験業務支援システム
に掲載されている同意説明文書の版数、作成日と実際に使用するために準備した同意説明文書の版
数、作成日の整合性を確認する。
なお、確認した時点で、旧版の同意説明文書は破棄する
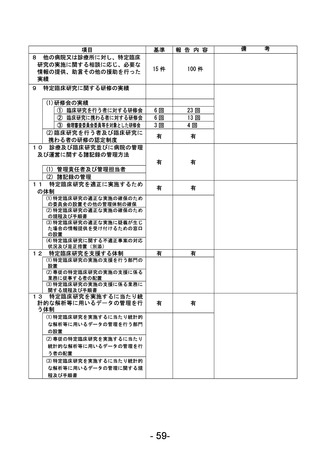
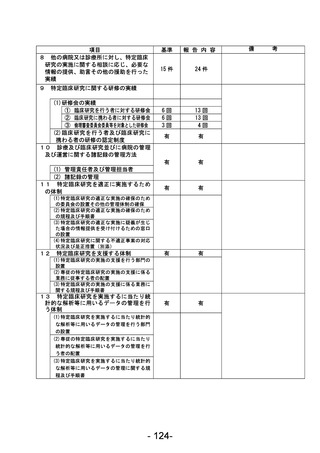
登録 ID 等
****
治験・臨床研究名
****
不適正事案の概要:
2024 年 1 月に変更申請において、研究期間 2024/3/31 から 2026/3/31、登録期間 2023/3/31 から
2025/3/31 を変更する際に、事務局確認において、2023/3/31 までの登録期間の終了に対して、登録
期間外に同意取得・登録したことが判明した。
2024 年 2 月 CRB にて登録期間外に同意取得した件について、被験者の権利保護にかかわるという点
も踏まえ、本件を重大として考えているか否かについて、研究代表医師に見解を求めたところ、重大
な不適合報告として提出する旨となった。
不適正事案に関する対応状況:
変更申請を提出。本件に参加頂いた患者の研究参加のご意思も鑑み、当該症例には「研究期間内では
あったが登録期間外の登録となった」旨をご説明の上、引き続き登録症例として経過観察を継続する
旨とした。
是正措置:
登録期間終了が研究期間終了よりも 1 年早いという認識が欠如していたこと、毎回の症例登録の際
に登録期間の確認を行っていなかったことが理由と考えられる。再発防止策としては、登録期間の認
識を改めて徹底するとともに、該当する時期に登録終了日の再確認および延長申請の必要性をリマ
インドさせるメールが届くように設定した。
CRB としては総括報告書を提出する際には、全症例の解析データとは別途、登録期間外に登録した症
例を除外した解析データも記載することもコメントとして挙げている。
(注)1
不適正事案に関する調査の概要(方法、期間、結果等)を記載すること。
- 89-