






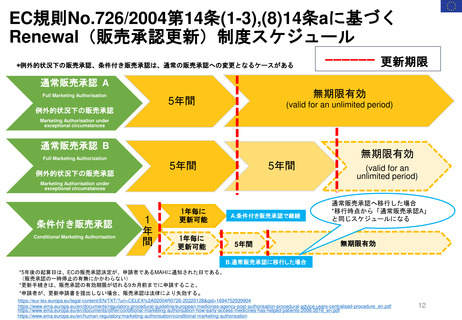
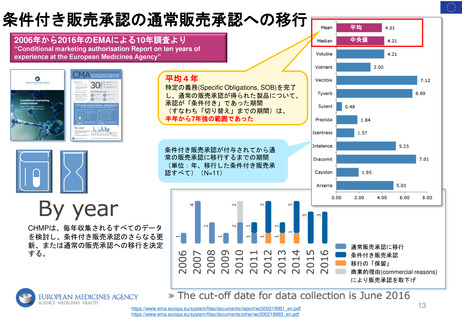
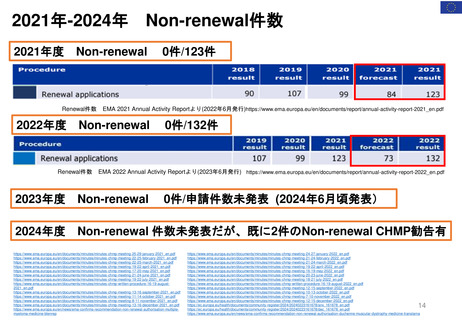

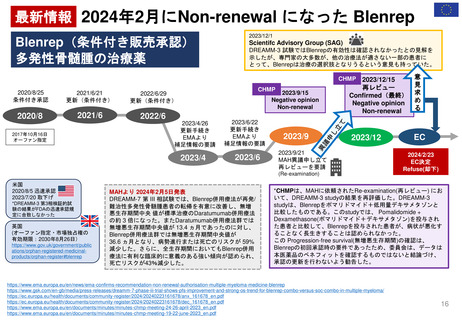











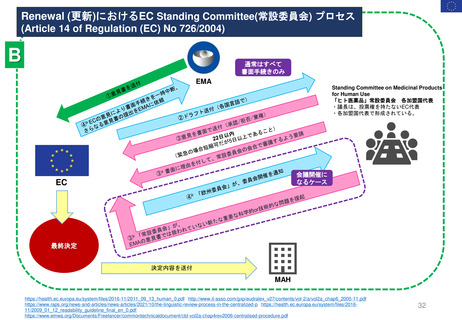

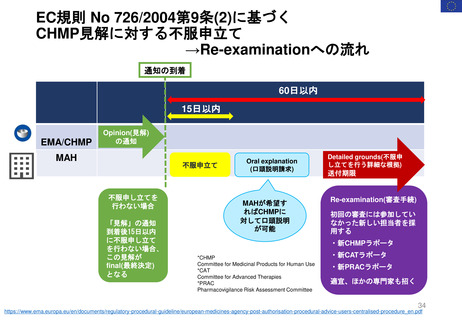
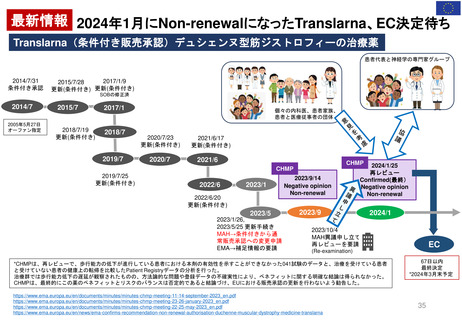

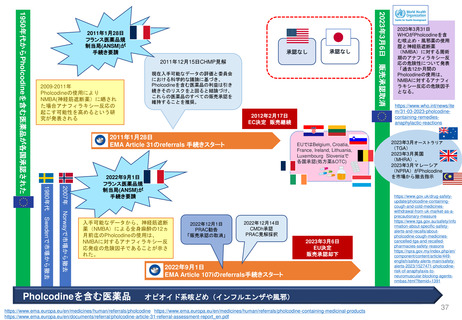



よむ、つかう、まなぶ。
【資料3】令和5年度欧米の市販後安全対策を中心とした薬事制度に関する調査[3.1MB] (24 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_38915.html |
| 出典情報 | 医薬品等行政評価・監視委員会(第15回 3/22)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
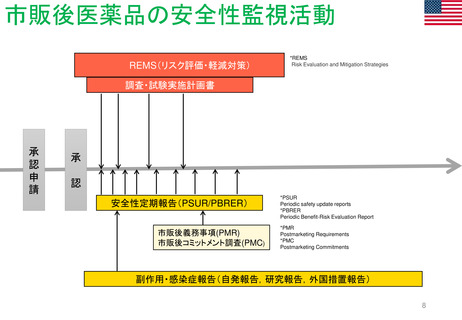
Postmarketing Requirements (PMR 市販後義務事項)
Postmarketing Commitments (PMC 市販後コミットメント調査)
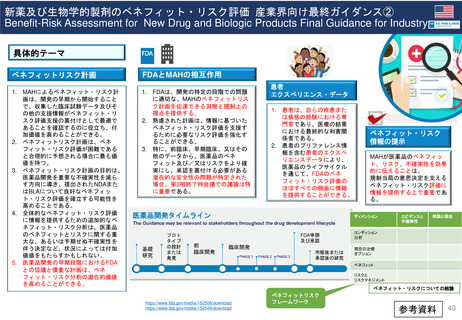
PMR及びPMCと臨床試験は、医薬品または生物学的製剤がFDAによって販売承認された後に行われる。 様々な法令や規制当局の下、FDAは特定の
MAHに対し、市販後試験や臨床試験の実施を要求することができる。 FDA改革法(2007年)では、特にFDAに医薬品に関連する可能性のある重大なリ
スクを評価するために、MAHに対してPMRと臨床試験の実施を要求する権限を与えている。
https://www.fda.gov/drugs/postmarket-requirements-and-commitments/postmarketing-requirements-and-commitments-reports
PMR
申請者が承認後に実施することを、法令または規則によ
り要求される Study(研究)または 臨床試験(Clinical trial)
のことである。
PMC
申請者が承認後に実施することに文書で同意する研究または臨床
試験のことであるが、法令または規則により義務付けられている
ものではない。
PMR、PMC関連の主要レポート、
ガイダンスなど
主な内容
1
The Food and Drug Administration Report on
the Performance of Drugs and Biologics Firms
in Conducting Postmarketing Requirements
and Commitments Fiscal Year 2021
https://www.fda.gov/media/165114/download
PMRとPMCを区別し、医薬品に関連する既知の重大なリスクを評価する上での重要性を強調している。本
文でFederal Food, Drug, and Cosmetic Act(FD&C法)第506B条に基づく年次報告義務を果たし、FDA内
部のPMR/PMCデータベースのデータの要約を提供、申請者に対する定期報告要件を述べている。
PMCを含む、市販後研究に関するMAHへの規制要件を詳述し、FD&C法が要求するPMR/PMC提出に関
するMAHの遵守状況を報告するFDAの役割を説明する。
オンライン検索、ダウンロード可能なデータベースは四半期ごとに更新され、報告書のステータス情報は毎年
更新される。このデータベースは全てのPMR/PMCのサブセットを含み、Openまたは過去1年以内にCloseし
た市販後研究と臨床試験のみを反映している。
2
Guidance for Industry, Postmarketing Studies
and Clinical Trials — Implementation of
Section 505(o)(3) of the Federal Food, Drug,
and Cosmetic Act,2011
https://www.fda.gov/media/131980/download
承認された処方薬及び生物学的製剤の市販後研究及び臨床試験をFDAに義務付ける権限を与えるFD&C
法第505条(o)(3)の実施について概説したものである。この文書は、MAHがFDAに要求される市販後調査及
び臨床試験に関する情報を、完了までのスケジュールも含めて規定している。第505条(o)(3)により義務付け
られた市販後研究及び臨床試験の要件を遵守することの重要性を強調している。FDA改革法2007が新たに
設けた 「研究」と 「臨床試験」の区別、FDA改革法が導入した権限と要件を強調している。
3
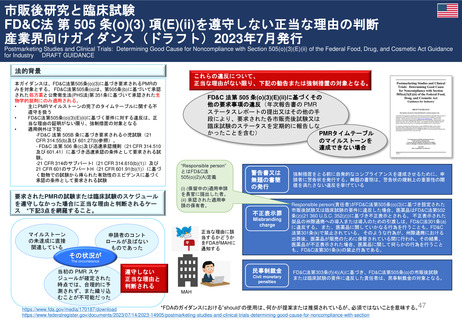
Postmarketing Studies and Clinical Trials—
Implementation of Section 505(o)(3) of the
Federal Food, Drug, and Cosmetic Act,
Guidance for Industry 2019(Draft)
https://www.fda.gov/media/148646/download
2番と同じ法律、条文に言及しているが、このガイダンスではFAERS(FDA Adverse Event Reporting
System)とVAERS(Vaccine Adverse Event Reporting System)の充足性に特に言及しており、一般的に
periodic safety reportsに含まれる情報を包含している。
また、法第505条(o)(3)の実施と、MAHがFDAに提供しなければならない情報を含む市販後研究及び臨床
試験の具体的な要件に焦点を当てている。
4
PDUFA REAUTHORIZATION
PERFORMANCE GOALS AND
PROCEDURES FISCAL YEARS
2023 THROUGH 2027
https://www.fda.gov/media/151712/download
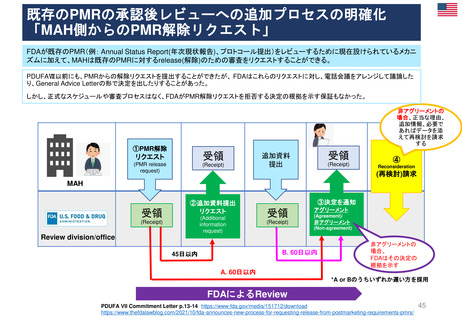
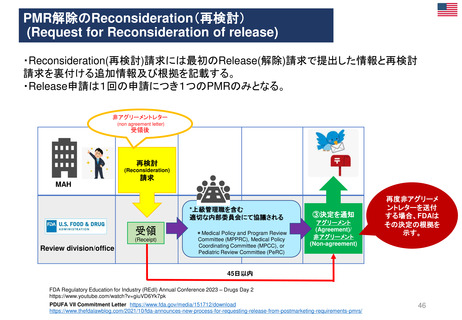
PDUFA VIIでは、PMRに関する新たなプロセス、スケジュール、及び実績目標が導入された。
承認後の既存のPMRのレビューのための新しいプロセスを導入し、予想されるPMR及びPMCについて
MAHと協議を継続し、試験または臨床試験のデザインを検討マイルストーンのスケジュールの提出を要求す
る。またMAH主導のPMR解除要求を審査する新しいプロセスの概要が示された。
24
Postmarketing Commitments (PMC 市販後コミットメント調査)
PMR及びPMCと臨床試験は、医薬品または生物学的製剤がFDAによって販売承認された後に行われる。 様々な法令や規制当局の下、FDAは特定の
MAHに対し、市販後試験や臨床試験の実施を要求することができる。 FDA改革法(2007年)では、特にFDAに医薬品に関連する可能性のある重大なリ
スクを評価するために、MAHに対してPMRと臨床試験の実施を要求する権限を与えている。
https://www.fda.gov/drugs/postmarket-requirements-and-commitments/postmarketing-requirements-and-commitments-reports
PMR
申請者が承認後に実施することを、法令または規則によ
り要求される Study(研究)または 臨床試験(Clinical trial)
のことである。
PMC
申請者が承認後に実施することに文書で同意する研究または臨床
試験のことであるが、法令または規則により義務付けられている
ものではない。
PMR、PMC関連の主要レポート、
ガイダンスなど
主な内容
1
The Food and Drug Administration Report on
the Performance of Drugs and Biologics Firms
in Conducting Postmarketing Requirements
and Commitments Fiscal Year 2021
https://www.fda.gov/media/165114/download
PMRとPMCを区別し、医薬品に関連する既知の重大なリスクを評価する上での重要性を強調している。本
文でFederal Food, Drug, and Cosmetic Act(FD&C法)第506B条に基づく年次報告義務を果たし、FDA内
部のPMR/PMCデータベースのデータの要約を提供、申請者に対する定期報告要件を述べている。
PMCを含む、市販後研究に関するMAHへの規制要件を詳述し、FD&C法が要求するPMR/PMC提出に関
するMAHの遵守状況を報告するFDAの役割を説明する。
オンライン検索、ダウンロード可能なデータベースは四半期ごとに更新され、報告書のステータス情報は毎年
更新される。このデータベースは全てのPMR/PMCのサブセットを含み、Openまたは過去1年以内にCloseし
た市販後研究と臨床試験のみを反映している。
2
Guidance for Industry, Postmarketing Studies
and Clinical Trials — Implementation of
Section 505(o)(3) of the Federal Food, Drug,
and Cosmetic Act,2011
https://www.fda.gov/media/131980/download
承認された処方薬及び生物学的製剤の市販後研究及び臨床試験をFDAに義務付ける権限を与えるFD&C
法第505条(o)(3)の実施について概説したものである。この文書は、MAHがFDAに要求される市販後調査及
び臨床試験に関する情報を、完了までのスケジュールも含めて規定している。第505条(o)(3)により義務付け
られた市販後研究及び臨床試験の要件を遵守することの重要性を強調している。FDA改革法2007が新たに
設けた 「研究」と 「臨床試験」の区別、FDA改革法が導入した権限と要件を強調している。
3
Postmarketing Studies and Clinical Trials—
Implementation of Section 505(o)(3) of the
Federal Food, Drug, and Cosmetic Act,
Guidance for Industry 2019(Draft)
https://www.fda.gov/media/148646/download
2番と同じ法律、条文に言及しているが、このガイダンスではFAERS(FDA Adverse Event Reporting
System)とVAERS(Vaccine Adverse Event Reporting System)の充足性に特に言及しており、一般的に
periodic safety reportsに含まれる情報を包含している。
また、法第505条(o)(3)の実施と、MAHがFDAに提供しなければならない情報を含む市販後研究及び臨床
試験の具体的な要件に焦点を当てている。
4
PDUFA REAUTHORIZATION
PERFORMANCE GOALS AND
PROCEDURES FISCAL YEARS
2023 THROUGH 2027
https://www.fda.gov/media/151712/download
PDUFA VIIでは、PMRに関する新たなプロセス、スケジュール、及び実績目標が導入された。
承認後の既存のPMRのレビューのための新しいプロセスを導入し、予想されるPMR及びPMCについて
MAHと協議を継続し、試験または臨床試験のデザインを検討マイルストーンのスケジュールの提出を要求す
る。またMAH主導のPMR解除要求を審査する新しいプロセスの概要が示された。
24