






























よむ、つかう、まなぶ。
資料3-6 コルヒチンの安全対策のための製造販売承認事項一部変更承認について[1.7MB] (18 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_73884.html |
| 出典情報 | 薬事審議会 医薬品等安全対策部会(令和8年度第1回 6/18)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
1066
TERKELTAUB ET AL
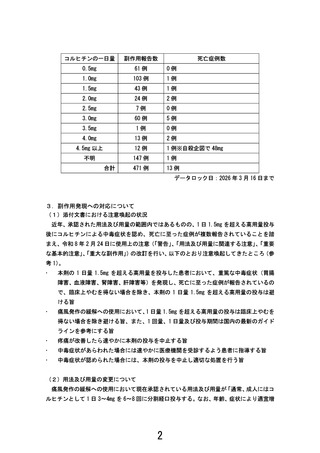
dose colchicine in a patient population likely to use
multiple concomitant drugs adds a safety margin without
compromising efficacy. Further studies are warranted to
optimize colchicine dosing with selected concomitant
medications.
The AGREE trial was designed to evaluate the
efficacy and safety of colchicine as typically administered in community practice. As such, colchicine was
self-administered within 12 hours of symptom onset, and
this study shows that therapy may be optimized by
employing the low-dose regimen described herein. Additional studies are needed to determine whether treatment start time, concomitant urate-lowering therapy,
extent of tophaceous crystal deposits, sites of arthritis,
renal function, or other doses affect efficacy or safety.
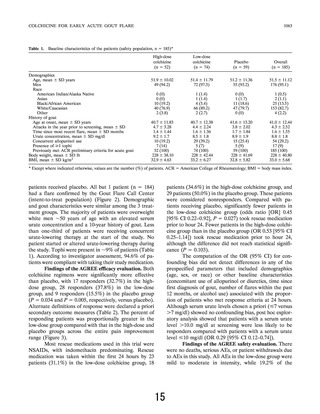
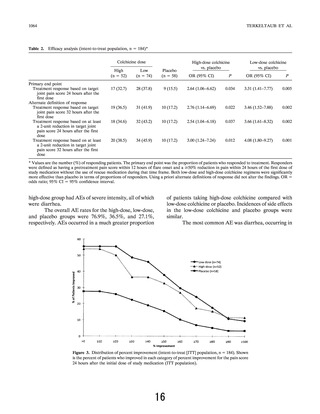
In the AGREE trial, the primary end point was
ⱖ50% pain reduction in 24 hours. A 50% reduction of
baseline pain, which is considered clinically significant
(20–22), was used in the original colchicine efficacy trial
(1) and continues to be a standard for measuring efficacy
in current acute gout studies (23). Patients who took
rescue medication within the first 24 hours after taking
the study drug, regardless of their intent, were not
classified as responders. In acute gout trials with
NSAIDs (24–26) or prednisone (26), the primary end
point has been therapy for ⱖ48 hours. Confounding
concomitant use of colchicine was allowed in some of
these trials (24,25), with some investigators not specifying its use. Regardless, the typical pain response of acute
gout to NSAIDs does not reach complete resolution
within the first 72 hours; in general, pain is reduced from
excruciating to bearable. For example, in a recent trial
comparing etoricoxib with indomethacin, the mean pain
reduction from maximum was only ⬃30% at 24 hours,
and 50% pain reduction was achieved only at 48–
72 hours (25).
Comparisons between the AGREE trial results
and those from previous gout trials with NSAIDs, corticosteroids, or high-dose colchicine (1,24–28) are limited by
several factors. Specifically, direct comparisons are precluded by dissimilar patient cohorts, allowing confounding concomitant medication, treatment randomized and
given in a research center rather than prerandomization
to self-treatment regimens, different end points, and
longer durations of the active gout flare prior to treatment than the 12-hour time point assessed in the
AGREE trial. Nevertheless, low-dose colchicine treatment may have advantages over NSAIDs or steroids in
certain populations, such as gout patients with renal,
gastrointestinal, or endocrine comorbidities that disfavor or prevent the use of these medications. A random-
ized, placebo-controlled trial comparing colchicine,
corticosteroids, and NSAIDs would be valuable. The
possibility that low-dose colchicine therapy may be
enhanced with concomitant use of corticosteroids,
NSAIDs, or other agents also needs to be explored.
All patients in this trial previously met the ACR
preliminary criteria for the classification of the acute
arthritis of primary gout (9). Our study focused on the
first 24 hours of a single gout flare. A critical feature of
our design was the attempt to overcome the ethical
issues of using a placebo control by limiting the time (1
day) that a patient in the placebo group would be
untreated. A 7-day active comparator, non–placebocontrolled trial against NSAIDs and corticosteroids
would allow comparisons with other studied treatment
regimens (24–26). This proposed active comparator,
non–placebo-controlled design would also give adequate
time to evaluate changes from baseline in quality of life,
joint function, physician global assessments, and effectiveness at days 2–7. Additional colchicine studies are
needed to determine whether concomitant uratelowering therapy, extent of tophaceous crystal deposits,
location of gout flare, renal function, or other dose
regimens affect efficacy or safety.
In conclusion, the results of the AGREE trial
provide the first evidence basis, after centuries of colchicine use, for low-dose colchicine therapy in the
treatment of early acute gout flare. The low-dose colchicine regimen maintained efficacy equivalent to that of
high-dose colchicine. It had a side effect profile significantly better than that of high-dose colchicine and
comparable with that of placebo. The results are consistent with recent, expert opinion–based European
League Against Rheumatism recommendations (8) and
support an immediate change in clinical practice from a
high-dose colchicine regimen to a low-dose colchicine
regimen for treatment of early gout flare.
ACKNOWLEDGMENTS
The authors wish to acknowledge the following contributors to this article: Joyce Sands (United BioSource Corporation), study leader; Kimberly Stulir (URL Pharma), study
management; Dianne Barry, PhD (Barry Medical Communications, LLC), Dr. Suman Wason (URL Pharma), and Dr.
Thomas Lauterio (URL Pharma), editing of final manuscript.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it
critically for important intellectual content, and all authors approved
the final version to be published. Dr. Terkeltaub had full access to all
18
TERKELTAUB ET AL
dose colchicine in a patient population likely to use
multiple concomitant drugs adds a safety margin without
compromising efficacy. Further studies are warranted to
optimize colchicine dosing with selected concomitant
medications.
The AGREE trial was designed to evaluate the
efficacy and safety of colchicine as typically administered in community practice. As such, colchicine was
self-administered within 12 hours of symptom onset, and
this study shows that therapy may be optimized by
employing the low-dose regimen described herein. Additional studies are needed to determine whether treatment start time, concomitant urate-lowering therapy,
extent of tophaceous crystal deposits, sites of arthritis,
renal function, or other doses affect efficacy or safety.
In the AGREE trial, the primary end point was
ⱖ50% pain reduction in 24 hours. A 50% reduction of
baseline pain, which is considered clinically significant
(20–22), was used in the original colchicine efficacy trial
(1) and continues to be a standard for measuring efficacy
in current acute gout studies (23). Patients who took
rescue medication within the first 24 hours after taking
the study drug, regardless of their intent, were not
classified as responders. In acute gout trials with
NSAIDs (24–26) or prednisone (26), the primary end
point has been therapy for ⱖ48 hours. Confounding
concomitant use of colchicine was allowed in some of
these trials (24,25), with some investigators not specifying its use. Regardless, the typical pain response of acute
gout to NSAIDs does not reach complete resolution
within the first 72 hours; in general, pain is reduced from
excruciating to bearable. For example, in a recent trial
comparing etoricoxib with indomethacin, the mean pain
reduction from maximum was only ⬃30% at 24 hours,
and 50% pain reduction was achieved only at 48–
72 hours (25).
Comparisons between the AGREE trial results
and those from previous gout trials with NSAIDs, corticosteroids, or high-dose colchicine (1,24–28) are limited by
several factors. Specifically, direct comparisons are precluded by dissimilar patient cohorts, allowing confounding concomitant medication, treatment randomized and
given in a research center rather than prerandomization
to self-treatment regimens, different end points, and
longer durations of the active gout flare prior to treatment than the 12-hour time point assessed in the
AGREE trial. Nevertheless, low-dose colchicine treatment may have advantages over NSAIDs or steroids in
certain populations, such as gout patients with renal,
gastrointestinal, or endocrine comorbidities that disfavor or prevent the use of these medications. A random-
ized, placebo-controlled trial comparing colchicine,
corticosteroids, and NSAIDs would be valuable. The
possibility that low-dose colchicine therapy may be
enhanced with concomitant use of corticosteroids,
NSAIDs, or other agents also needs to be explored.
All patients in this trial previously met the ACR
preliminary criteria for the classification of the acute
arthritis of primary gout (9). Our study focused on the
first 24 hours of a single gout flare. A critical feature of
our design was the attempt to overcome the ethical
issues of using a placebo control by limiting the time (1
day) that a patient in the placebo group would be
untreated. A 7-day active comparator, non–placebocontrolled trial against NSAIDs and corticosteroids
would allow comparisons with other studied treatment
regimens (24–26). This proposed active comparator,
non–placebo-controlled design would also give adequate
time to evaluate changes from baseline in quality of life,
joint function, physician global assessments, and effectiveness at days 2–7. Additional colchicine studies are
needed to determine whether concomitant uratelowering therapy, extent of tophaceous crystal deposits,
location of gout flare, renal function, or other dose
regimens affect efficacy or safety.
In conclusion, the results of the AGREE trial
provide the first evidence basis, after centuries of colchicine use, for low-dose colchicine therapy in the
treatment of early acute gout flare. The low-dose colchicine regimen maintained efficacy equivalent to that of
high-dose colchicine. It had a side effect profile significantly better than that of high-dose colchicine and
comparable with that of placebo. The results are consistent with recent, expert opinion–based European
League Against Rheumatism recommendations (8) and
support an immediate change in clinical practice from a
high-dose colchicine regimen to a low-dose colchicine
regimen for treatment of early gout flare.
ACKNOWLEDGMENTS
The authors wish to acknowledge the following contributors to this article: Joyce Sands (United BioSource Corporation), study leader; Kimberly Stulir (URL Pharma), study
management; Dianne Barry, PhD (Barry Medical Communications, LLC), Dr. Suman Wason (URL Pharma), and Dr.
Thomas Lauterio (URL Pharma), editing of final manuscript.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it
critically for important intellectual content, and all authors approved
the final version to be published. Dr. Terkeltaub had full access to all
18