
















よむ、つかう、まなぶ。
【資料1】医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律等の一部を改正する法律の施行に向けた論点等について.pdf (20 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_59820.html |
| 出典情報 | 厚生科学審議会 医薬品医療機器制度部会(令和7年度第2回 7/23)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
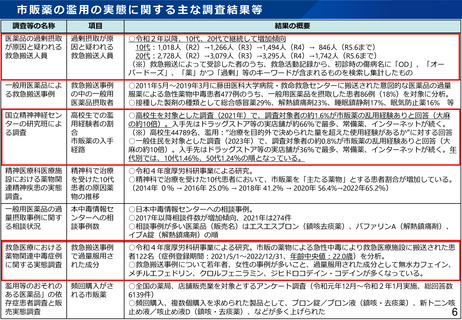
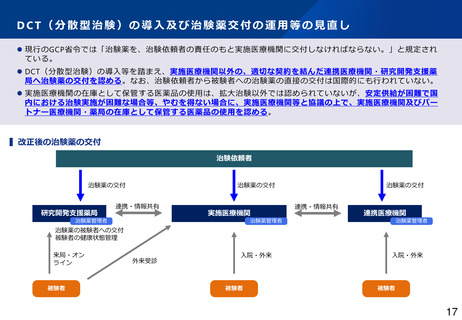
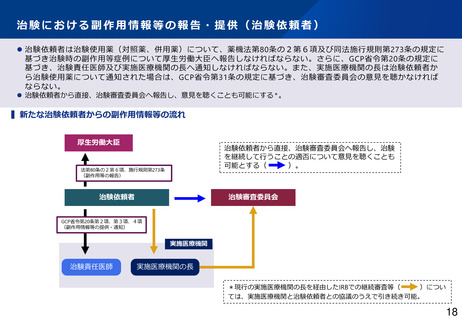
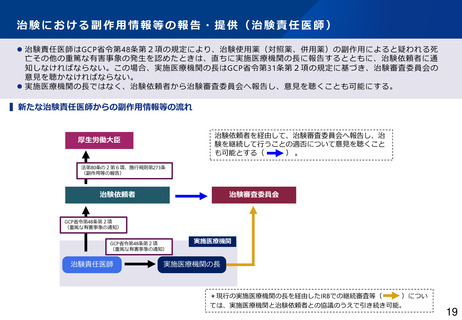
治験における副作用等情報の収集・評価体制について
▍治験における副作用等情報の情報源
海外市販後情報
国内治験・海外臨床試験情報
⚫ 企業(海外製販・国内依頼者)への報告主体
⚫ 依頼者への報告主体
⚫ 得られる情報の質
⚫ 得られる情報の質
• 医療従事者
• 患者等の非医療従事者
• SNS、ウェブサイト等の情報
• 情報が不確かで、因果関係が相当低いとみられるものも多数
• これらの情報は治験依頼者から、全て「未知」の有害事象として
規制当局及び実施医療機関に報告される。
必ずしも十分でない情報が、規制当局や実施医療機関に対して極めて短期
間に個別症例報告が行われている。
(規制当局:7日/15日以内 実施医療機関:直ちに)
⚫
• 医療機関にとっても有益な情報が得られる。
• 治験責任医師により臨床的に評価され、治験依頼者から規制当局
及び実施医療機関に報告される。
(参考)PMDAへの薬物の治験中の副作用等報告件数
(課題)
⚫
海外の試験情報は、精
密であり、引き続き報
告を求める
• 治験責任医師
膨大な件数の症例報告により、実施医療機関等の負担が増大しており、重
要な情報が埋もれる恐れがある。
情報源
平成31年度
令和2年度
令和3年度
令和4年度
令和5年度
国内
1,296
1,088
1,142
1,177
1,104
海外
93,570
133,823
251,951
168,051
164,678
➢ 海外情報の大部分は海外市販後情報。
➢ 上記報告のうち、「未知」の症例が実施医療機関への報告対象。
▍海外の規制
⚫
EUではEU域外における海外市販後情報は報告対象外*1。アメリカにおいて海外市販後情報を報告対象外とする直接の規定はないが、因果関係が合
理的に認められる場合に報告対象が限定され、因果関係が不明確な症例についてFDA及び治験実施医療機関へ報告は行われない*2。
⚫
ICH-E6(R3) において、依頼者による安全性情報の収集及び評価が規定され、因果関係が合理的に認められる情報を規制当局及び治験実施医療機関へ
伝達することが要求されている*3。
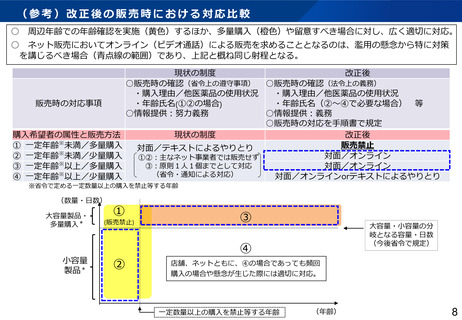
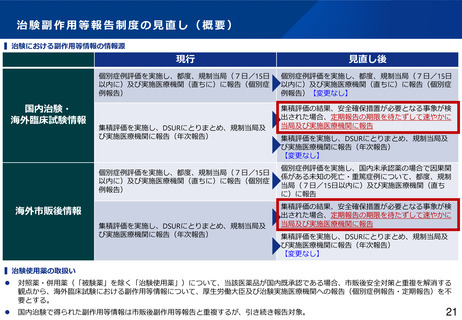
▍見直しの方向性(案)
⚫
海外市販後情報の報告対象を見直す。集積評価により、治験の中止・中断、プロトコル・治験薬概要書の改訂等の安全確保措置が必要となる事象が
検出された場合、定期報告の期限を待たずして速やかに厚生労働省及び実施医療機関等への報告対象とする。
*1
*2
*3
Detailed guidance on the collection, verification and presentation of adverse event/reaction reports arising from clinical trials on medicinal products for human use (‘CT-3’) (2011/C 172/01)
Guidance for Industry and Investigators Safety Reporting Requirements for INDs and BA/BE Studies (HHS,FDA CDER, CBER, December 2012)
ICH E6 (R3) Guideline for good clinical practice (GCP)
20
▍治験における副作用等情報の情報源
海外市販後情報
国内治験・海外臨床試験情報
⚫ 企業(海外製販・国内依頼者)への報告主体
⚫ 依頼者への報告主体
⚫ 得られる情報の質
⚫ 得られる情報の質
• 医療従事者
• 患者等の非医療従事者
• SNS、ウェブサイト等の情報
• 情報が不確かで、因果関係が相当低いとみられるものも多数
• これらの情報は治験依頼者から、全て「未知」の有害事象として
規制当局及び実施医療機関に報告される。
必ずしも十分でない情報が、規制当局や実施医療機関に対して極めて短期
間に個別症例報告が行われている。
(規制当局:7日/15日以内 実施医療機関:直ちに)
⚫
• 医療機関にとっても有益な情報が得られる。
• 治験責任医師により臨床的に評価され、治験依頼者から規制当局
及び実施医療機関に報告される。
(参考)PMDAへの薬物の治験中の副作用等報告件数
(課題)
⚫
海外の試験情報は、精
密であり、引き続き報
告を求める
• 治験責任医師
膨大な件数の症例報告により、実施医療機関等の負担が増大しており、重
要な情報が埋もれる恐れがある。
情報源
平成31年度
令和2年度
令和3年度
令和4年度
令和5年度
国内
1,296
1,088
1,142
1,177
1,104
海外
93,570
133,823
251,951
168,051
164,678
➢ 海外情報の大部分は海外市販後情報。
➢ 上記報告のうち、「未知」の症例が実施医療機関への報告対象。
▍海外の規制
⚫
EUではEU域外における海外市販後情報は報告対象外*1。アメリカにおいて海外市販後情報を報告対象外とする直接の規定はないが、因果関係が合
理的に認められる場合に報告対象が限定され、因果関係が不明確な症例についてFDA及び治験実施医療機関へ報告は行われない*2。
⚫
ICH-E6(R3) において、依頼者による安全性情報の収集及び評価が規定され、因果関係が合理的に認められる情報を規制当局及び治験実施医療機関へ
伝達することが要求されている*3。
▍見直しの方向性(案)
⚫
海外市販後情報の報告対象を見直す。集積評価により、治験の中止・中断、プロトコル・治験薬概要書の改訂等の安全確保措置が必要となる事象が
検出された場合、定期報告の期限を待たずして速やかに厚生労働省及び実施医療機関等への報告対象とする。
*1
*2
*3
Detailed guidance on the collection, verification and presentation of adverse event/reaction reports arising from clinical trials on medicinal products for human use (‘CT-3’) (2011/C 172/01)
Guidance for Industry and Investigators Safety Reporting Requirements for INDs and BA/BE Studies (HHS,FDA CDER, CBER, December 2012)
ICH E6 (R3) Guideline for good clinical practice (GCP)
20