









よむ、つかう、まなぶ。
総-7参考2 (5 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_71462.html |
| 出典情報 | 中央社会保険医療協議会 総会(第648回 3/11)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
3.臨床成績
気管支喘息(既存治療によっても喘息症状をコントロールできない重症又は難治の患者に
限る)の承認時に評価を行った主な臨床試験の成績を示す。
国際共同第Ⅲ相試験(EFC13579 試験)(成人及び 12 歳以上の小児)
【試験の概要】
中用量又は高用量の吸入ステロイド薬(以下、「ICS」)及びその他の長期管理薬を使用し
てもコントロール不良な 12 歳以上の気管支喘息患者 1,902 例(日本人 114 例を含む)を対象
に、
ICS 及びその他の長期管理薬1)1~2 剤併用下での本剤の有効性及び安全性を検討するため、
プラセボ対照無作為化二重盲検並行群間比較試験が実施された。
用法・用量は、本剤 200 mg(初回のみ 400 mg)、300 mg(初回のみ 600 mg)又はプラセボ
を 2 週間隔で 52 週間皮下投与することと設定され、ICS 及びその他の長期管理薬 1~2 剤をス
クリーニング時に確認された用量で併用することと設定された。
有効性の主要評価項目は、投与 52 週後までの重度喘息増悪2)の年間発現率及び投与 12 週後
における気管支拡張薬投与前の FEV1 のベースラインからの変化量の co-primary endpoint と設
定された。
対象となる患者は、12 歳以上の気管支喘息患者で、以下の基準を満たすこととされた。
(主な選択基準)
➢
中用量又は高用量の ICS3)及び長期管理薬 1~2 剤をスクリーニング時の 3 カ月以上前か
ら使用かつスクリーニング時の 1 カ月以上前から一定用量で継続して使用している
➢
気管支拡張薬投与前の FEV1 が予測値の 80%以下(17 歳以下は 90%以下)
➢
ACQ-5 スコアが 1.5 以上
➢
サルブタモール 200~400 µg 投与後の FEV1 に 12%以上かつ改善量が 200 mL 以上の可逆
性が認められる
➢
1 年以内に喘息悪化に対して全身性ステロイド薬の投与を 1 回以上受けた又は喘息悪化に
より入院若しくは救急外来を受診した
【結果】
承認用量が投与された本剤 300 mg/2 mL 群(以下、「本剤群」)と、解析に際して当該用量
群と対比較することとされたプラセボ/2 mL 群(以下、「プラセボ群」)の成績のみ提示する。
(有効性)
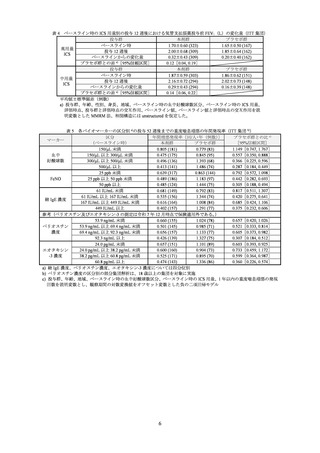
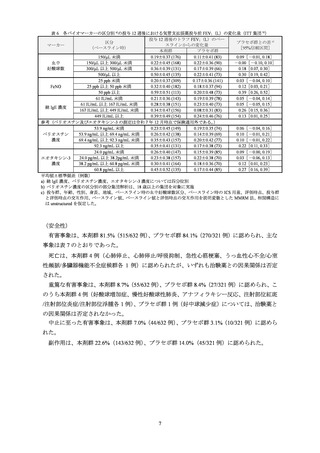
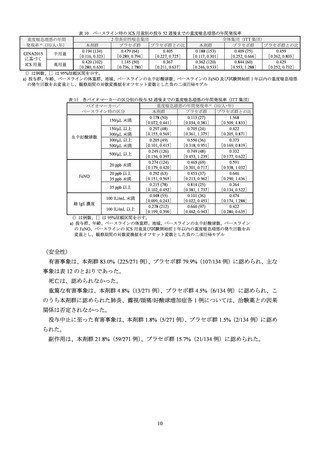
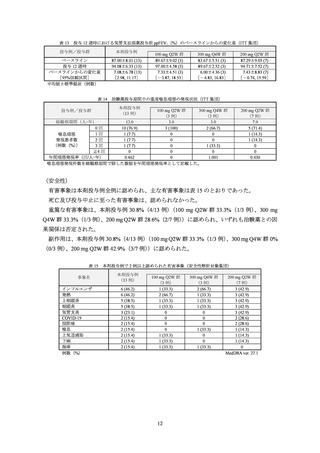
有効性の主要評価項目である投与 52 週後までの重度喘息増悪の年間発現率及び投与 12 週
後における気管支拡張薬投与前の FEV1 のベースラインからの変化量は表 1 及び表 2 のとおり
であり、プラセボ群と本剤群との対比較において、両主要評価項目で共に統計学的な有意差が
認められた。
1)
長時間作用性 β2 刺激薬(以下、
「LABA」
)、ロイコトリエン受容体拮抗薬(以下、
「LTRA」
)、長時間作用性抗コリン薬(以
下、「LAMA」)
、メチルキサンチン類
2)
次の①又は②の対応が必要な喘息の悪化を重度喘息増悪と定義した:①全身ステロイド薬の 3 日間以上の投与、②全身ス
テロイド薬の投与が必要な喘息による入院又は救急外来の受診
3)
フルチカゾンプロピオン酸エステル(以下、
「FP」)500 μg/日以上 2,000 μg/日以下相当。本邦からの被験者では、18 歳以
上は FP 400 μg/日以上 2,000 μg/日以下相当、17 歳以下は FP 200 μg/日以上 2,000 μg/日以下相当とされた。なお、本邦にお
ける FP の承認用量は、成人で最大 800 μg/日、小児で最大 200 μg/日である。
4
気管支喘息(既存治療によっても喘息症状をコントロールできない重症又は難治の患者に
限る)の承認時に評価を行った主な臨床試験の成績を示す。
国際共同第Ⅲ相試験(EFC13579 試験)(成人及び 12 歳以上の小児)
【試験の概要】
中用量又は高用量の吸入ステロイド薬(以下、「ICS」)及びその他の長期管理薬を使用し
てもコントロール不良な 12 歳以上の気管支喘息患者 1,902 例(日本人 114 例を含む)を対象
に、
ICS 及びその他の長期管理薬1)1~2 剤併用下での本剤の有効性及び安全性を検討するため、
プラセボ対照無作為化二重盲検並行群間比較試験が実施された。
用法・用量は、本剤 200 mg(初回のみ 400 mg)、300 mg(初回のみ 600 mg)又はプラセボ
を 2 週間隔で 52 週間皮下投与することと設定され、ICS 及びその他の長期管理薬 1~2 剤をス
クリーニング時に確認された用量で併用することと設定された。
有効性の主要評価項目は、投与 52 週後までの重度喘息増悪2)の年間発現率及び投与 12 週後
における気管支拡張薬投与前の FEV1 のベースラインからの変化量の co-primary endpoint と設
定された。
対象となる患者は、12 歳以上の気管支喘息患者で、以下の基準を満たすこととされた。
(主な選択基準)
➢
中用量又は高用量の ICS3)及び長期管理薬 1~2 剤をスクリーニング時の 3 カ月以上前か
ら使用かつスクリーニング時の 1 カ月以上前から一定用量で継続して使用している
➢
気管支拡張薬投与前の FEV1 が予測値の 80%以下(17 歳以下は 90%以下)
➢
ACQ-5 スコアが 1.5 以上
➢
サルブタモール 200~400 µg 投与後の FEV1 に 12%以上かつ改善量が 200 mL 以上の可逆
性が認められる
➢
1 年以内に喘息悪化に対して全身性ステロイド薬の投与を 1 回以上受けた又は喘息悪化に
より入院若しくは救急外来を受診した
【結果】
承認用量が投与された本剤 300 mg/2 mL 群(以下、「本剤群」)と、解析に際して当該用量
群と対比較することとされたプラセボ/2 mL 群(以下、「プラセボ群」)の成績のみ提示する。
(有効性)
有効性の主要評価項目である投与 52 週後までの重度喘息増悪の年間発現率及び投与 12 週
後における気管支拡張薬投与前の FEV1 のベースラインからの変化量は表 1 及び表 2 のとおり
であり、プラセボ群と本剤群との対比較において、両主要評価項目で共に統計学的な有意差が
認められた。
1)
長時間作用性 β2 刺激薬(以下、
「LABA」
)、ロイコトリエン受容体拮抗薬(以下、
「LTRA」
)、長時間作用性抗コリン薬(以
下、「LAMA」)
、メチルキサンチン類
2)
次の①又は②の対応が必要な喘息の悪化を重度喘息増悪と定義した:①全身ステロイド薬の 3 日間以上の投与、②全身ス
テロイド薬の投与が必要な喘息による入院又は救急外来の受診
3)
フルチカゾンプロピオン酸エステル(以下、
「FP」)500 μg/日以上 2,000 μg/日以下相当。本邦からの被験者では、18 歳以
上は FP 400 μg/日以上 2,000 μg/日以下相当、17 歳以下は FP 200 μg/日以上 2,000 μg/日以下相当とされた。なお、本邦にお
ける FP の承認用量は、成人で最大 800 μg/日、小児で最大 200 μg/日である。
4