



















よむ、つかう、まなぶ。
資料1 医療用医薬品の安定供給確保に向けた取組について (13 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_65290.html |
| 出典情報 | 厚生科学審議会 医療用医薬品迅速・安定供給部会(第2回 10/27)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
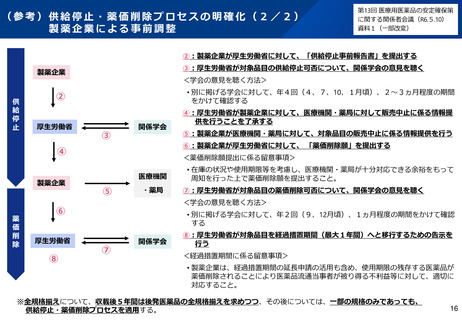
(参考)GMP適合性調査制度の見直しについて(令和7年改正薬機法)
改正の概要
<定期のGMP適合性調査の合理化>
•
リスクに応じてメリハリをつけた適合性調査を行う観点から、定期のGMP適合性調査について、調査頻度を見直す(※)と
ともに、調査権者が調査に先立ちリスク評価を行うこととして、製造管理・品質管理上のリスクが低いと評価した場合には調
査不要とし、リスクが高いと評価した製造所に対して重点的に実地調査を行えるようにする。
※定期のGMP適合性調査の調査頻度を5年ごと⇒3年ごとに短縮(政令改正にて対応予定)
<区分適合性調査の追加調査>
•
区分適合性調査について、製造工程区分が「製造管理又は品質管理に関し特に注意が必要な区分」に該当するときは、
都道府県に加え、PMDAも都道府県と協力して調査(追加調査)を行うこととする(※ただし、過去の調査結果等を勘
案してその必要がないと認める場合を除く)。
<基準確認証制度の拡大>
•
基準確認証により定期のGMP適合性調査を省略できる対象範囲を拡大し、グローバルサプライチェーンが複雑化・多様化
する中、国際整合の観点から、輸出用医薬品も基準確認証制度の対象とする。
•
製造方法等の中リスクの変更カテゴリの追加に伴い、変更のリスクが中程度である場合の一部変更の承認について、基準
確認証の交付を受けているときは、適合性調査の実施を不要とする。
<新規後発医薬品の調査主体移管>
•
後発医薬品(後発品として初めて承認を受ける成分を含有する品目に限る。)について、製造開始時における製造管
理・品質管理上の不備が発生するリスクが特に高いことを踏まえ、製剤工程に係る新規承認時の適合性調査の実施主体
を、都道府県からPMDAに見直す(政令改正にて対応予定)。
施行期日
•
公布後2年以内に政令で定める日(<基準確認証制度の拡大>の中リスク変更については、公布後3年以内に政令で定める日)
13
改正の概要
<定期のGMP適合性調査の合理化>
•
リスクに応じてメリハリをつけた適合性調査を行う観点から、定期のGMP適合性調査について、調査頻度を見直す(※)と
ともに、調査権者が調査に先立ちリスク評価を行うこととして、製造管理・品質管理上のリスクが低いと評価した場合には調
査不要とし、リスクが高いと評価した製造所に対して重点的に実地調査を行えるようにする。
※定期のGMP適合性調査の調査頻度を5年ごと⇒3年ごとに短縮(政令改正にて対応予定)
<区分適合性調査の追加調査>
•
区分適合性調査について、製造工程区分が「製造管理又は品質管理に関し特に注意が必要な区分」に該当するときは、
都道府県に加え、PMDAも都道府県と協力して調査(追加調査)を行うこととする(※ただし、過去の調査結果等を勘
案してその必要がないと認める場合を除く)。
<基準確認証制度の拡大>
•
基準確認証により定期のGMP適合性調査を省略できる対象範囲を拡大し、グローバルサプライチェーンが複雑化・多様化
する中、国際整合の観点から、輸出用医薬品も基準確認証制度の対象とする。
•
製造方法等の中リスクの変更カテゴリの追加に伴い、変更のリスクが中程度である場合の一部変更の承認について、基準
確認証の交付を受けているときは、適合性調査の実施を不要とする。
<新規後発医薬品の調査主体移管>
•
後発医薬品(後発品として初めて承認を受ける成分を含有する品目に限る。)について、製造開始時における製造管
理・品質管理上の不備が発生するリスクが特に高いことを踏まえ、製剤工程に係る新規承認時の適合性調査の実施主体
を、都道府県からPMDAに見直す(政令改正にて対応予定)。
施行期日
•
公布後2年以内に政令で定める日(<基準確認証制度の拡大>の中リスク変更については、公布後3年以内に政令で定める日)
13