











































よむ、つかう、まなぶ。
【資料4】モルヌピラビル(ラゲブリオカプセル200mg )の安全性について(医薬・生活衛生局の説明資料) (7 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_24538.html |
| 出典情報 | 医薬品等行政評価・監視委員会(第7回 3/18)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
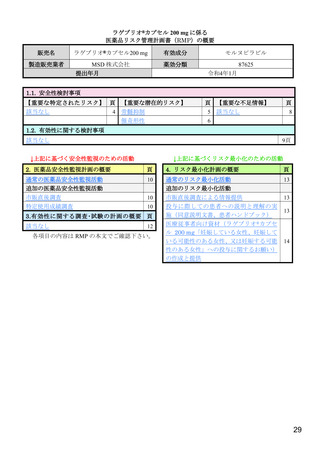
2.1
海外第Ⅱa 相試験(MK-4482-006 試験<2020 年 6 月~2021 年 2 月>)
18 歳以上の SARS-CoV-2 による感染症患者(目標例数 172 例6))を対象に、本薬の抗ウイルス活性及
び安全性を検討することを目的として、プラセボ対照無作為化二重盲検並行群間比較試験7)が、米国の
11 施設で実施された。本試験の主な選択・除外基準は表 1 のとおりであった。
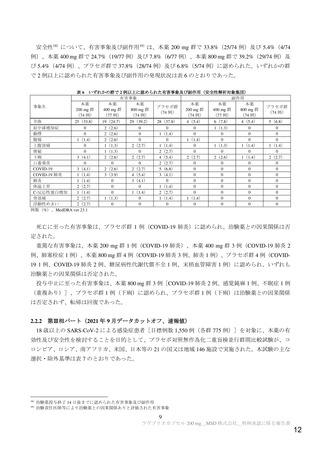
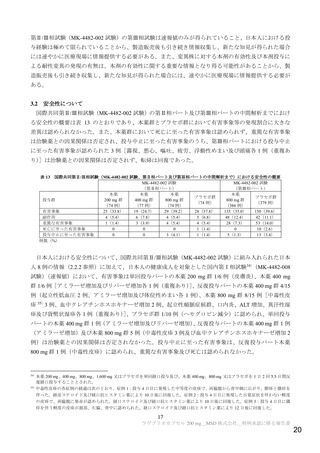
表 1 主な選択・除外基準
1. SARS-CoV-2 陽性(無作為化前 96 時間以内に採取された検体を用いた PCR 検査等により確認)
2. 症状発現から治験薬投与開始まで 168 時間以内 a)と見込まれる
選択基準
3. 無作為化時点において SARS-CoV-2 による感染症の症状(発熱又は呼吸器症状)b)が認められる
4. 治験薬投与開始から治験薬投与終了後 100 日間の避妊が可能な男性又は妊娠しておらず治験薬投与開始から治験薬
投与終了後 50 日間の避妊が可能な女性
1. 入院や早急な治療を要する
2. ヘモグロビン値が男性の場合 10 g/dL 未満、女性の場合 9 g/dL 未満
3. 血小板数 100,000/μL 未満 c)又は無作為化前 5 日以内に血小板輸血を受けた患者
4. 透析中又は eGFR が 30 mL/min/1.73 m2 未満 d)
5. AST 又は ALT が基準値上限の 3 倍超
除外基準
6. SARS-CoV-2 による感染症のために入院中又は入院歴を有する
7. 腎疾患の既往歴(クレアチニンクリアランスが 30 mL/min 未満 e))を有する
8. 肝疾患の既往歴又は活動性の HBV 又は HCV の現病を有する
9. CD4 が 200/mm3 未満又はヌクレオシドアナログで治療中の HIV 患者
10. SARS-CoV-2 による感染症に対するワクチンの接種歴を有する
a)プロトコル第 2 版(2020 年 7 月 22 日)で 120 時間以内から 168 時間以内に変更された。
b)発熱:熱っぽさ又は悪寒の訴えを含む。呼吸器症状:嗅覚消失、味覚消失、喉の痛み、咳又は息切れ等が含まれる。
c)プロトコル第 5 版(2020 年 11 月 15 日)で 125,000/μL 未満から 100,000/μL 未満に変更された。
d)プロトコル第 5 版(2020 年 11 月 15 日)で eGFR が 60 mL/min/1.73 m2 未満から 30 mL/min/1.73 m2 未満に変更された。
e)プロトコル第 5 版(2020 年 11 月 15 日)でクレアチニンクリアランスが 60 mL/min 未満から 30 mL/min 未満に変更された。
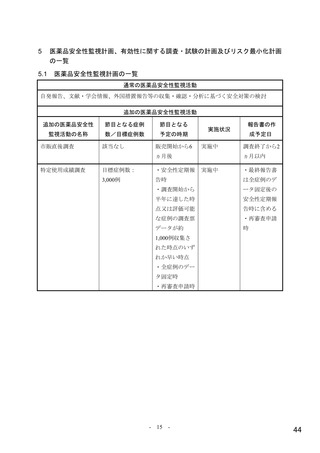
用法・用量は、本薬 200 mg、400 mg、800 mg 又はプラセボを 1 日 2 回、5 日間経口投与することとさ
れた。
無作為化された 204 例のうち、治験薬が 1 回以上投与された 202 例(本薬 200 mg 群 23 例、本薬 400 mg
群 62 例、本薬 800 mg 群 55 例、プラセボ群 62 例)が安全性解析対象集団であった。無作為化された 204
例のうち、治験薬が 1 回以上投与され、かつ、治験薬投与開始後の RT-PCR 検査結果(鼻咽頭ぬぐい検
体)が少なくとも 1 時点で得られた 198 例(本薬 200 mg 群 23 例、本薬 400 mg 群 61 例、本薬 800 mg 群
53 例、プラセボ群 61 例)が MITT 集団とされ、抗ウイルス活性の解析対象集団であった。
試験中止例は 7 例(本薬 400 mg 群 3 例、本薬 800 mg 群 3 例、プラセボ群 1 例)であり、中止理由の
内訳は、有害事象 3 例(本薬 400 mg 群 1 例、本薬 800 mg 群 1 例、プラセボ群 1 例)、追跡不能 1 例(本
薬 400 mg 群)、治験責任医師等の判断8)1 例(本薬 800 mg 群)、被験者の申し出 2 例(本薬 400 mg 群
1 例、本薬 800 mg 群 1 例)であった。
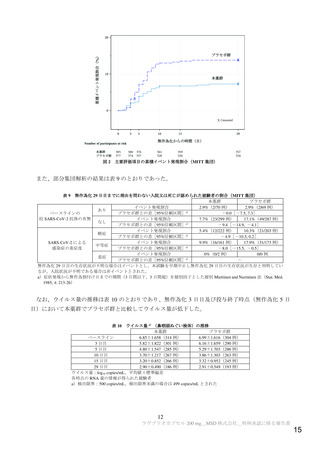
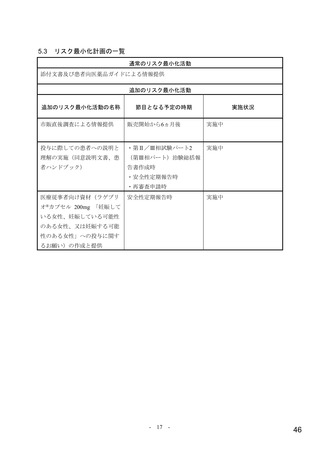
抗ウイルス活性について、主要評価項目である治験薬投与開始時点から RT-PCR 検査結果が陰性とな
った検体採取日までの時間9)は表 2 及び図 1 のとおりであった。
プロトコル第 1 版(2020 年 5 月 29 日)において、本薬 200 mg 又はプラセボを投与する計画[目標例数 44 例(各群 22 例)]として
試験が開始された。その後、プロトコル第 3 版(2020 年 8 月 27 日)において、本薬 800 mg を超えない範囲で本薬の高用量又はプラ
セボを投与する 4 パート(パート 2~5)が追加され、目標例数が 108 例[うち、パート 2~5:各 16 例(本薬群 12 例、プラセボ群 4
例)]に変更された。さらに、プロトコル第 5 版(2020 年 11 月 15 日)において、4 パート(パート 6~9)が追加され、目標例数が
172 例[うち、パート 6~9:各 16 例(本薬群 12 例、プラセボ群 4 例)]に変更され、被験者が治験を中止したことにより抗ウイルス
活性のデータが得られない場合等は新たな被験者が組み入れられるが全体の被験者数が 204 例を超えないこととされた。
7)
本試験に直接的に関与していないスポンサー内のメンバーは、用量追加等の判断を目的として非盲検下において抗ウイルス活性に関
する情報を評価可能であった。
8)
服薬コンプライアンスに関する懸念が理由とされた。
9)
鼻咽頭ぬぐい検体を用いた RT-PCR 検査において、SARS-CoV-2 の RNA 量が定量下限(1,018 copies/mL)未満の場合に陰性とされた。
また、RT-PCR 検査に用いる鼻咽頭ぬぐい検体は、治験薬投与開始前、無作為化 2、3、4、5、7、10、14、28 日目及び治験中止時に採
取することとされた。なお、プロトコル第 4 版(2020 年 9 月 10 日)において、無作為化 2、4 及び 10 日目の検体採取が削除された。
6)
4
ラゲブリオカプセル 200 mg_MSD 株式会社_特例承認に係る報告書
7
海外第Ⅱa 相試験(MK-4482-006 試験<2020 年 6 月~2021 年 2 月>)
18 歳以上の SARS-CoV-2 による感染症患者(目標例数 172 例6))を対象に、本薬の抗ウイルス活性及
び安全性を検討することを目的として、プラセボ対照無作為化二重盲検並行群間比較試験7)が、米国の
11 施設で実施された。本試験の主な選択・除外基準は表 1 のとおりであった。
表 1 主な選択・除外基準
1. SARS-CoV-2 陽性(無作為化前 96 時間以内に採取された検体を用いた PCR 検査等により確認)
2. 症状発現から治験薬投与開始まで 168 時間以内 a)と見込まれる
選択基準
3. 無作為化時点において SARS-CoV-2 による感染症の症状(発熱又は呼吸器症状)b)が認められる
4. 治験薬投与開始から治験薬投与終了後 100 日間の避妊が可能な男性又は妊娠しておらず治験薬投与開始から治験薬
投与終了後 50 日間の避妊が可能な女性
1. 入院や早急な治療を要する
2. ヘモグロビン値が男性の場合 10 g/dL 未満、女性の場合 9 g/dL 未満
3. 血小板数 100,000/μL 未満 c)又は無作為化前 5 日以内に血小板輸血を受けた患者
4. 透析中又は eGFR が 30 mL/min/1.73 m2 未満 d)
5. AST 又は ALT が基準値上限の 3 倍超
除外基準
6. SARS-CoV-2 による感染症のために入院中又は入院歴を有する
7. 腎疾患の既往歴(クレアチニンクリアランスが 30 mL/min 未満 e))を有する
8. 肝疾患の既往歴又は活動性の HBV 又は HCV の現病を有する
9. CD4 が 200/mm3 未満又はヌクレオシドアナログで治療中の HIV 患者
10. SARS-CoV-2 による感染症に対するワクチンの接種歴を有する
a)プロトコル第 2 版(2020 年 7 月 22 日)で 120 時間以内から 168 時間以内に変更された。
b)発熱:熱っぽさ又は悪寒の訴えを含む。呼吸器症状:嗅覚消失、味覚消失、喉の痛み、咳又は息切れ等が含まれる。
c)プロトコル第 5 版(2020 年 11 月 15 日)で 125,000/μL 未満から 100,000/μL 未満に変更された。
d)プロトコル第 5 版(2020 年 11 月 15 日)で eGFR が 60 mL/min/1.73 m2 未満から 30 mL/min/1.73 m2 未満に変更された。
e)プロトコル第 5 版(2020 年 11 月 15 日)でクレアチニンクリアランスが 60 mL/min 未満から 30 mL/min 未満に変更された。
用法・用量は、本薬 200 mg、400 mg、800 mg 又はプラセボを 1 日 2 回、5 日間経口投与することとさ
れた。
無作為化された 204 例のうち、治験薬が 1 回以上投与された 202 例(本薬 200 mg 群 23 例、本薬 400 mg
群 62 例、本薬 800 mg 群 55 例、プラセボ群 62 例)が安全性解析対象集団であった。無作為化された 204
例のうち、治験薬が 1 回以上投与され、かつ、治験薬投与開始後の RT-PCR 検査結果(鼻咽頭ぬぐい検
体)が少なくとも 1 時点で得られた 198 例(本薬 200 mg 群 23 例、本薬 400 mg 群 61 例、本薬 800 mg 群
53 例、プラセボ群 61 例)が MITT 集団とされ、抗ウイルス活性の解析対象集団であった。
試験中止例は 7 例(本薬 400 mg 群 3 例、本薬 800 mg 群 3 例、プラセボ群 1 例)であり、中止理由の
内訳は、有害事象 3 例(本薬 400 mg 群 1 例、本薬 800 mg 群 1 例、プラセボ群 1 例)、追跡不能 1 例(本
薬 400 mg 群)、治験責任医師等の判断8)1 例(本薬 800 mg 群)、被験者の申し出 2 例(本薬 400 mg 群
1 例、本薬 800 mg 群 1 例)であった。
抗ウイルス活性について、主要評価項目である治験薬投与開始時点から RT-PCR 検査結果が陰性とな
った検体採取日までの時間9)は表 2 及び図 1 のとおりであった。
プロトコル第 1 版(2020 年 5 月 29 日)において、本薬 200 mg 又はプラセボを投与する計画[目標例数 44 例(各群 22 例)]として
試験が開始された。その後、プロトコル第 3 版(2020 年 8 月 27 日)において、本薬 800 mg を超えない範囲で本薬の高用量又はプラ
セボを投与する 4 パート(パート 2~5)が追加され、目標例数が 108 例[うち、パート 2~5:各 16 例(本薬群 12 例、プラセボ群 4
例)]に変更された。さらに、プロトコル第 5 版(2020 年 11 月 15 日)において、4 パート(パート 6~9)が追加され、目標例数が
172 例[うち、パート 6~9:各 16 例(本薬群 12 例、プラセボ群 4 例)]に変更され、被験者が治験を中止したことにより抗ウイルス
活性のデータが得られない場合等は新たな被験者が組み入れられるが全体の被験者数が 204 例を超えないこととされた。
7)
本試験に直接的に関与していないスポンサー内のメンバーは、用量追加等の判断を目的として非盲検下において抗ウイルス活性に関
する情報を評価可能であった。
8)
服薬コンプライアンスに関する懸念が理由とされた。
9)
鼻咽頭ぬぐい検体を用いた RT-PCR 検査において、SARS-CoV-2 の RNA 量が定量下限(1,018 copies/mL)未満の場合に陰性とされた。
また、RT-PCR 検査に用いる鼻咽頭ぬぐい検体は、治験薬投与開始前、無作為化 2、3、4、5、7、10、14、28 日目及び治験中止時に採
取することとされた。なお、プロトコル第 4 版(2020 年 9 月 10 日)において、無作為化 2、4 及び 10 日目の検体採取が削除された。
6)
4
ラゲブリオカプセル 200 mg_MSD 株式会社_特例承認に係る報告書
7