

































































よむ、つかう、まなぶ。
【資料No.1】2.5_臨床に関する概括資料 (128 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_29325.html |
| 出典情報 | 薬事・食品衛生審議会 薬事分科会(令和4年度第5回 11/22)、医薬品第二部会(令和4年度第13回 11/22)(合同開催)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
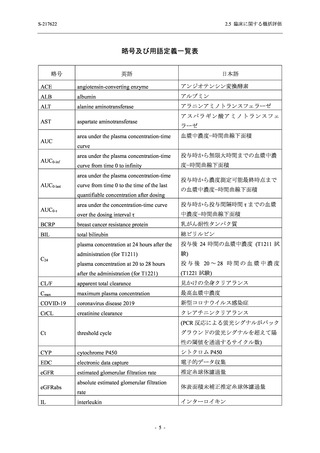
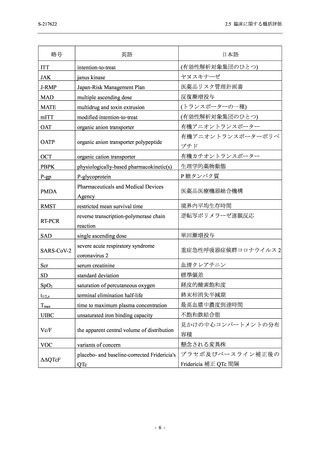
S-217622
2.5 臨床に関する概括評価
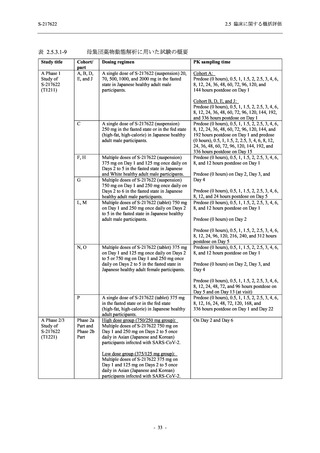
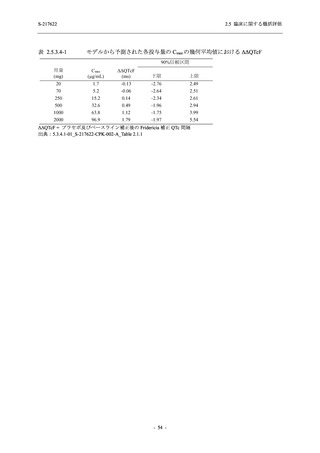
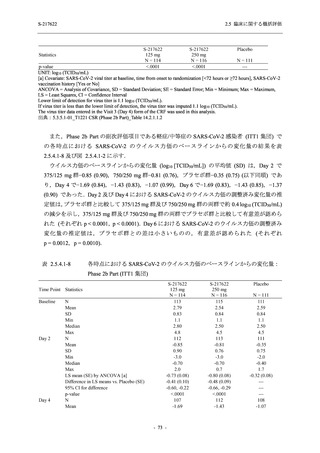
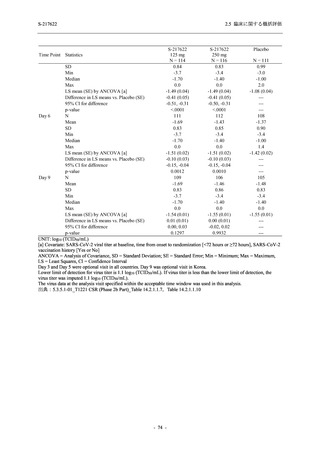
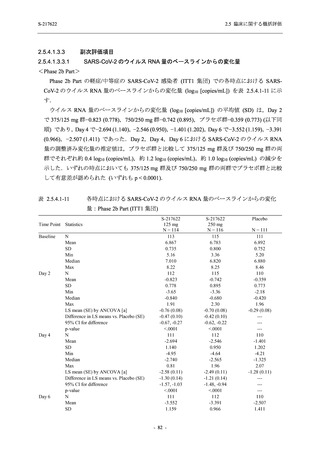
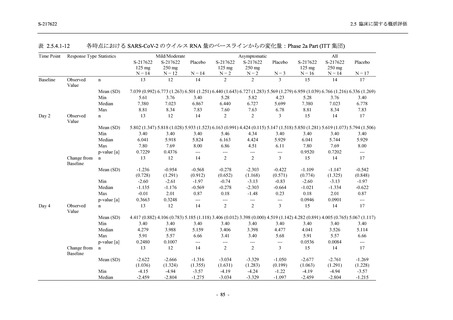
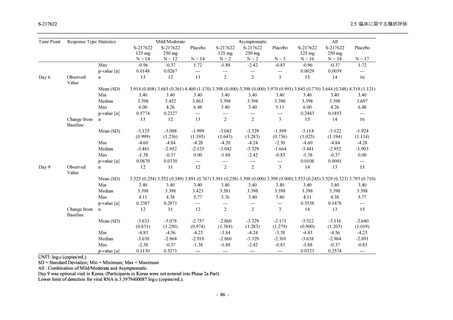
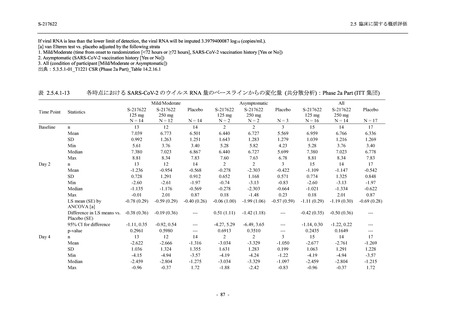
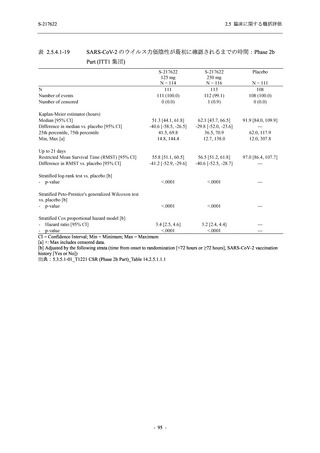
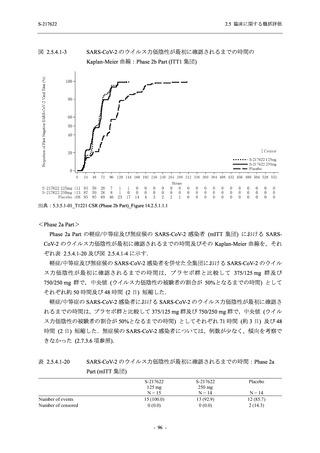
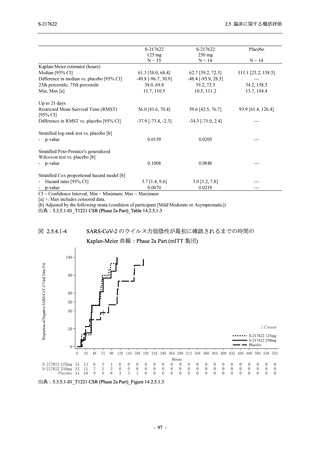
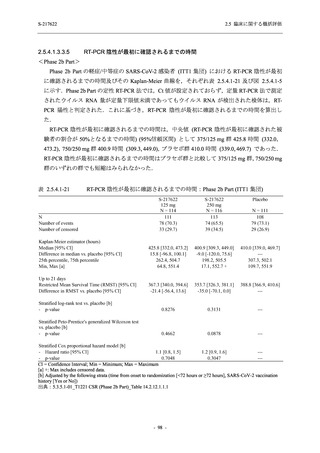
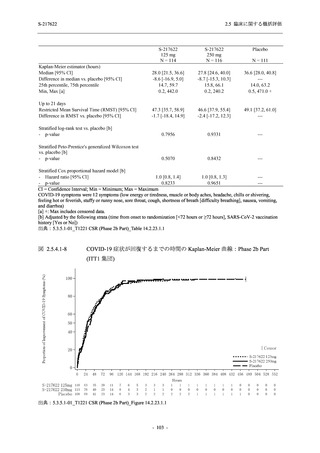
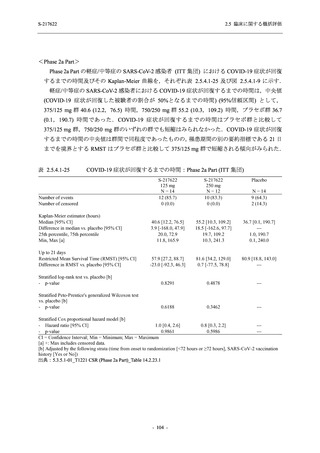
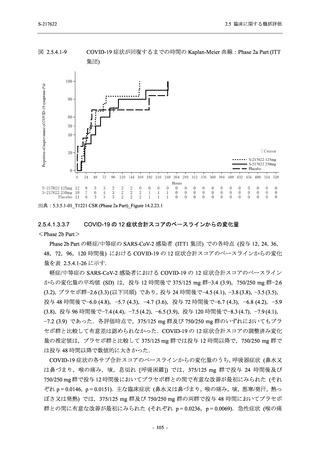
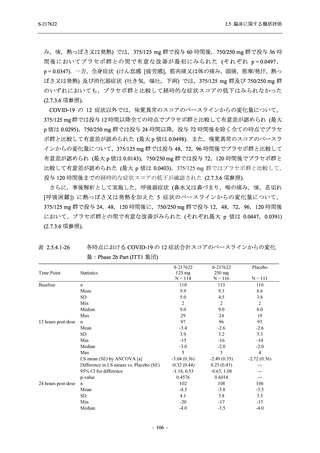
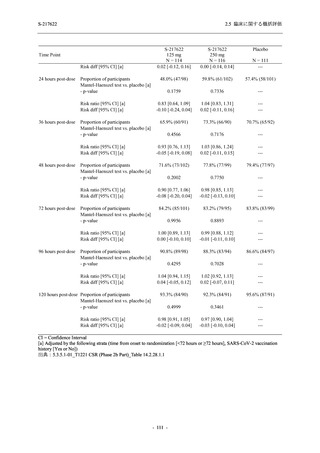
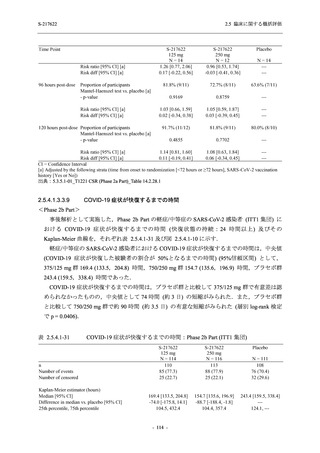
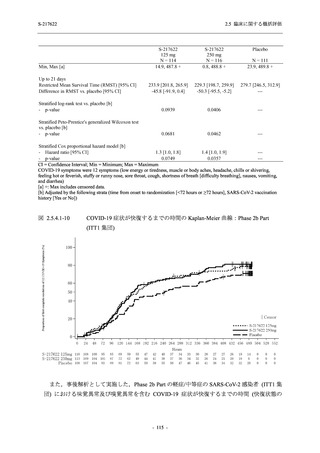
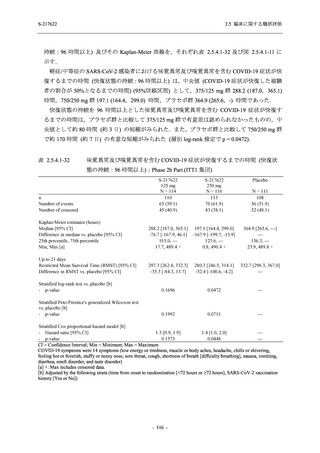
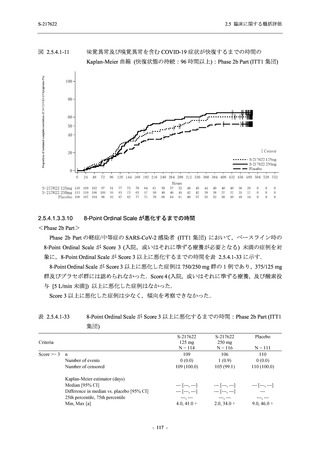
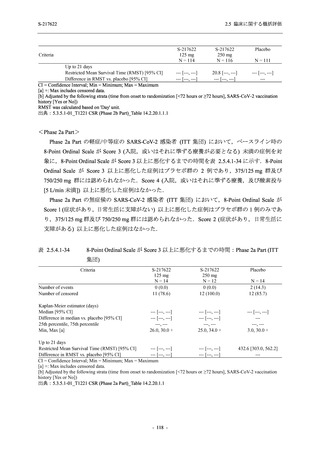
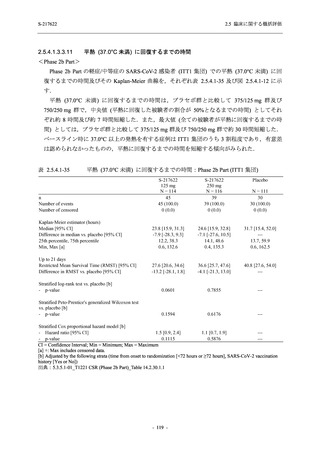
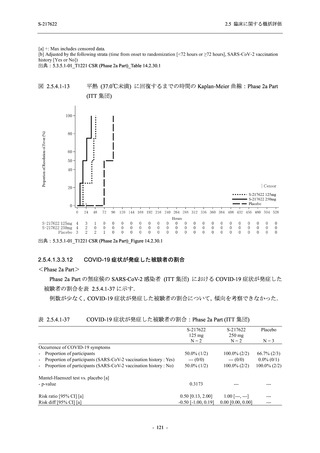
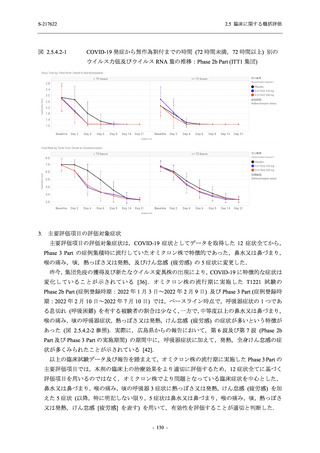
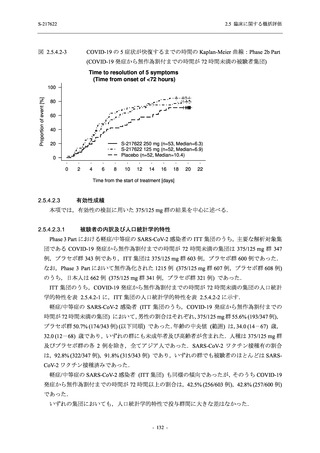
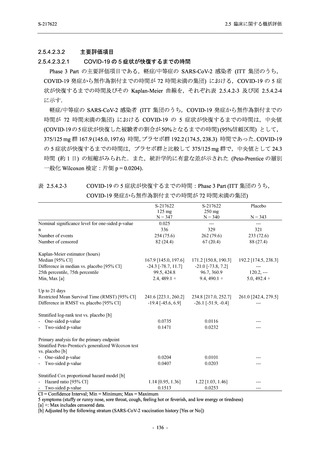
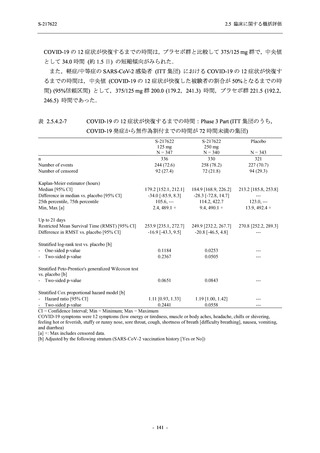
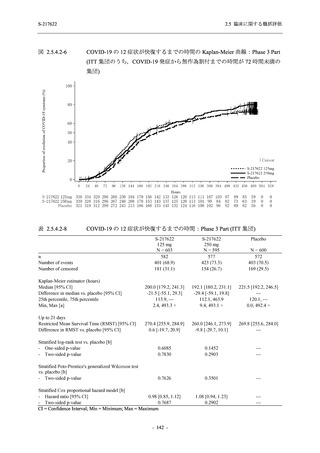
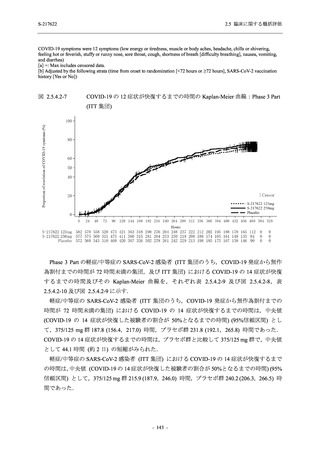
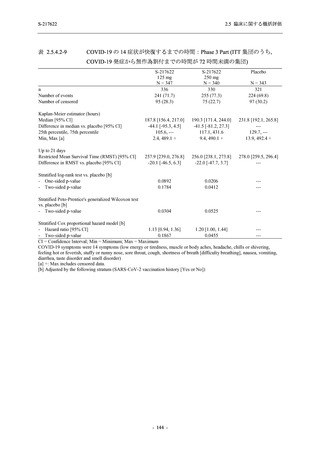
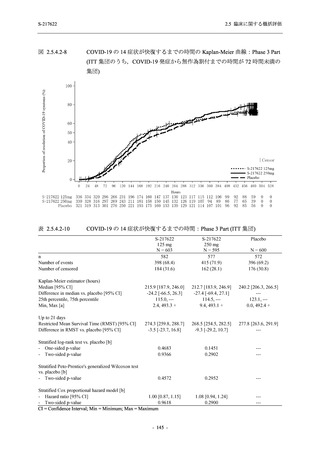
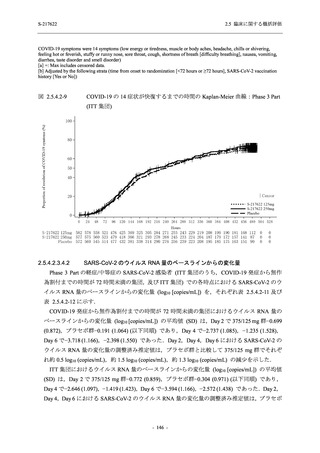
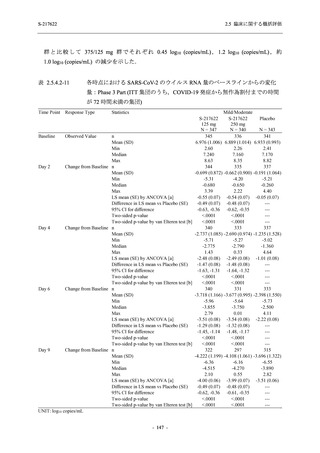
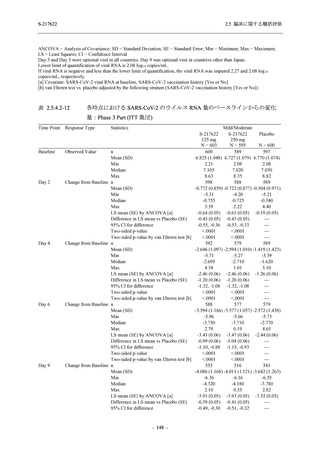
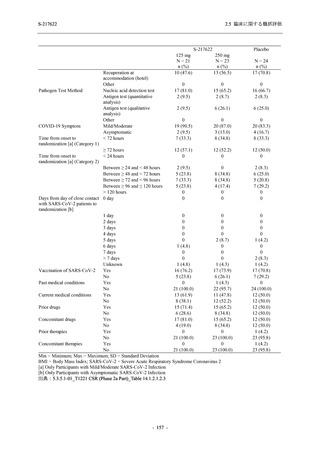
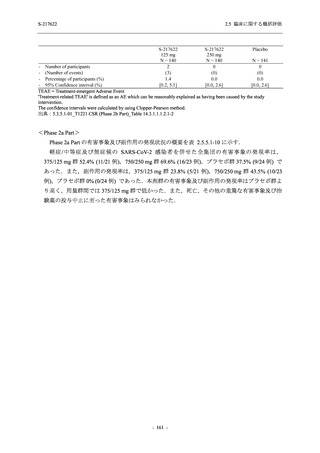
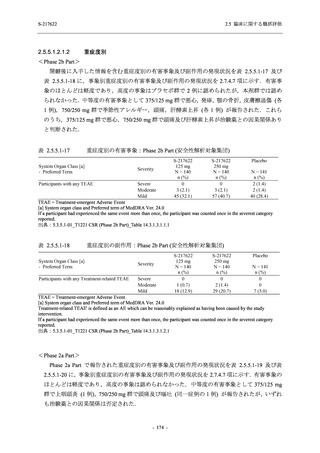
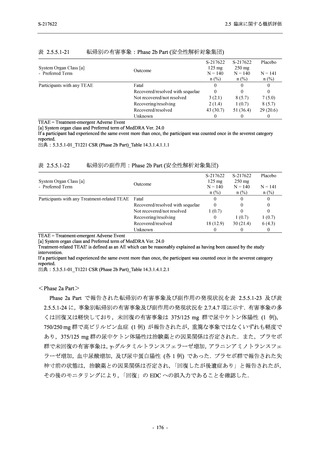
また,Phase 3 Part の副次評価項目として,COVID-19 の 14 症状が快復するまでの時間も評価
し,本評価項目に対して,COVID-19 の 5 症状が快復するまでの時間と同様の解析を行った.
COVID-19 の 14 症状の「快復」は,Phase 3 Part の主要評価項目に記載した「快復」に加え,
治験薬投与開始時点から味覚異常及び嗅覚異常が下記の通りに快復した時点を指し,その状態
が少なくとも 24 時間持続したとき,COVID-19 の 14 症状が快復したと判断した.
●
COVID-19 発症前から存在した既存症状で,ベースライン (投与前検査) 時点で悪化し
ていると被験者が判断した症状については,ベースライン時点の重症度から改善又は
維持する必要がある.
●
COVID-19 発症前から存在した既存症状で,ベースライン (投与前検査) 時点で悪化し
ていないと被験者が判断した症状については,ベースライン時点の重症度を維持又は
改善する必要がある.
●
上記以外の症状,すなわち COVID-19 発症前には存在しておらず,ベースライン (投与
前検査) 時点あるいはそれ以降に発現した症状については,症状が「通常通り」になる
必要がある.
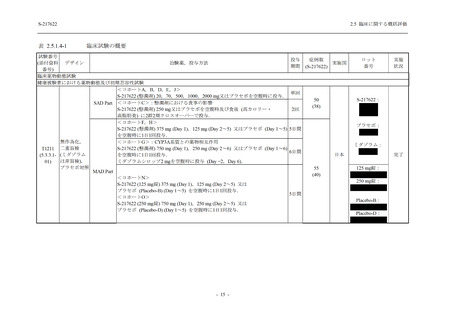
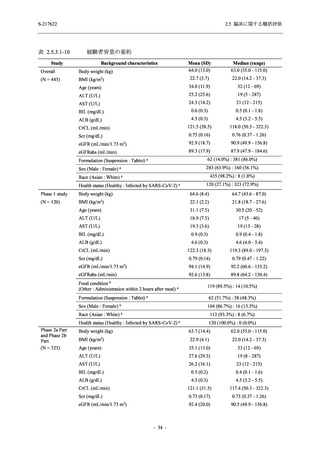
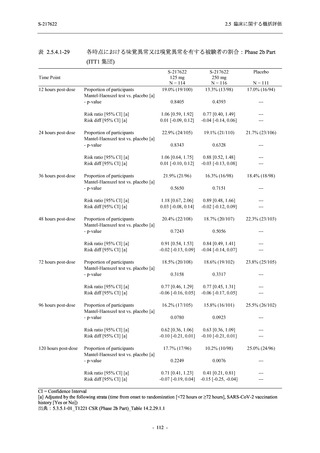
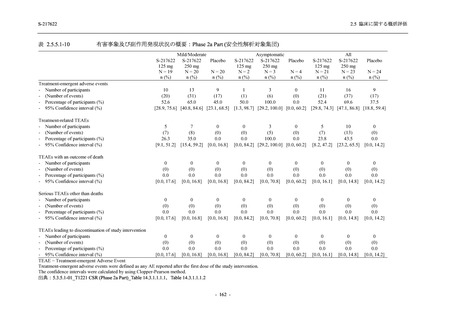
2.5.4.2.2.3
T1221 試験 Phase 3 Part の計画変更内容
2.5.1.3 項で前述した Phase 3 Part の計画変更 (治験実施計画書第 10 版) について,主な変更
内容及びその妥当性を以下に示す.
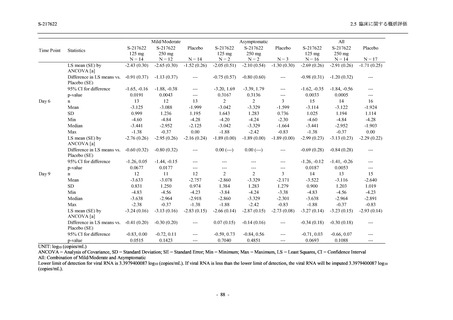
1. 有効性の検証対象の用量
Phase 3 Part では,有効性の検証対象の用量を 375/125 mg 群及び 750/250 mg 群の 2 群から,
375/125 mg 群のみに変更した.Phase 2b Part の結果を用いた薬物動態/薬力学解析から,両投与
量による曝露間で本剤の有効性に明確な差はなく,375/125 mg 群で十分な抗ウイルス効果が確
認できたことから [39],本剤の申請用法・用量として低用量である 375/125 mg を選択すること
は妥当と判断した.高用量である 750/250 mg 群については,安全性解析及び副次的な位置付け
としての有効性解析を実施することとした.
なお,本剤の申請用法用量を 375/125 mg として,2022 年 2 月 25 日に製造承認販売申請を行
い,同年 7 月 20 日の薬事分科会・医薬品第二部会 (合同開催) において,Phase 3 Part 等の結果
をもって継続審議と判断されたが,承認される場合,用法用量を 375/125 mg とすることは妥当
と判断されている [39].
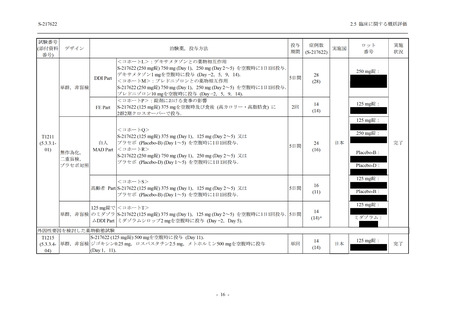
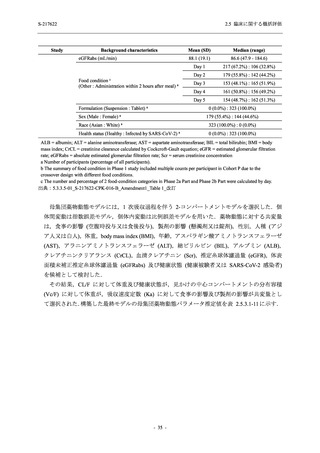
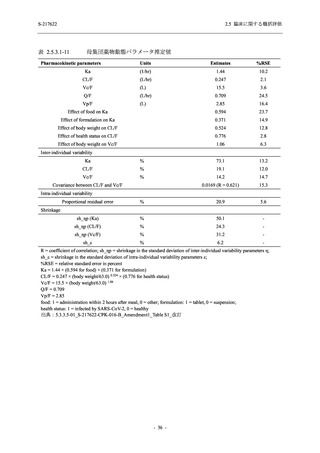
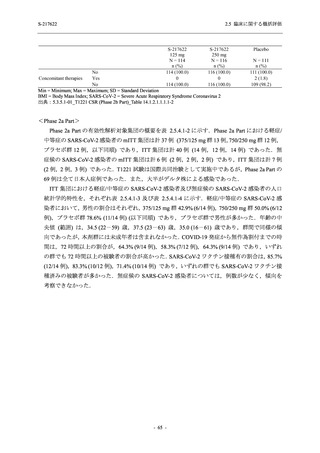
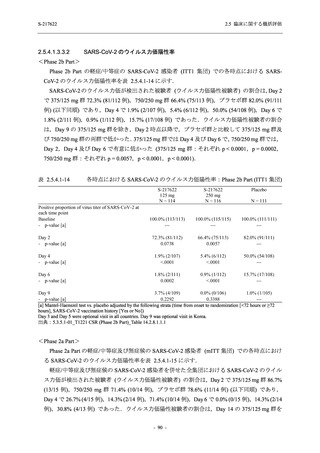
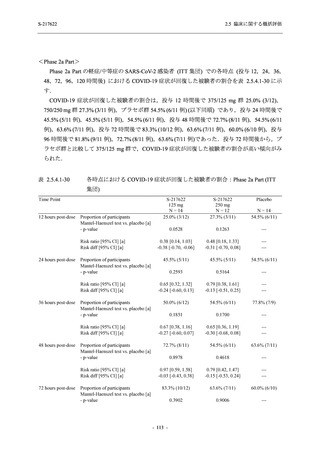
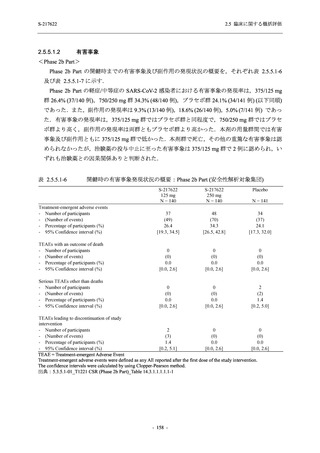
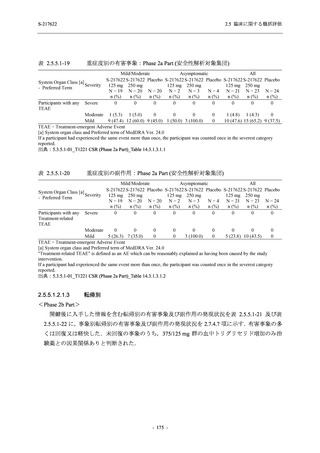
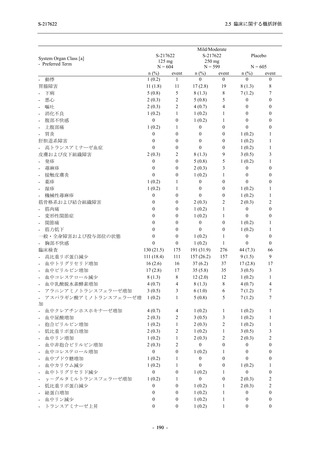
2. 主要解析の解析対象集団
主要評価項目及び主要な副次評価項目を対象とした主要解析における多重性調整の固定順序
法において,まず ITT 集団のうち,COVID-19 発症から無作為割付までの時間が 72 時間未満の
被験者に限定した解析対象集団で 375/125 mg 群とプラセボ群の比較を目的とした検定を行うこ
ととした.ここで有意な差が認められた場合には,引き続き,COVID-19 発症から無作為割付ま
での時間が 120 時間以内の被験者を対象とした ITT 集団全体を対象として検定を行うこととし
た.
- 128 -
2.5 臨床に関する概括評価
また,Phase 3 Part の副次評価項目として,COVID-19 の 14 症状が快復するまでの時間も評価
し,本評価項目に対して,COVID-19 の 5 症状が快復するまでの時間と同様の解析を行った.
COVID-19 の 14 症状の「快復」は,Phase 3 Part の主要評価項目に記載した「快復」に加え,
治験薬投与開始時点から味覚異常及び嗅覚異常が下記の通りに快復した時点を指し,その状態
が少なくとも 24 時間持続したとき,COVID-19 の 14 症状が快復したと判断した.
●
COVID-19 発症前から存在した既存症状で,ベースライン (投与前検査) 時点で悪化し
ていると被験者が判断した症状については,ベースライン時点の重症度から改善又は
維持する必要がある.
●
COVID-19 発症前から存在した既存症状で,ベースライン (投与前検査) 時点で悪化し
ていないと被験者が判断した症状については,ベースライン時点の重症度を維持又は
改善する必要がある.
●
上記以外の症状,すなわち COVID-19 発症前には存在しておらず,ベースライン (投与
前検査) 時点あるいはそれ以降に発現した症状については,症状が「通常通り」になる
必要がある.
2.5.4.2.2.3
T1221 試験 Phase 3 Part の計画変更内容
2.5.1.3 項で前述した Phase 3 Part の計画変更 (治験実施計画書第 10 版) について,主な変更
内容及びその妥当性を以下に示す.
1. 有効性の検証対象の用量
Phase 3 Part では,有効性の検証対象の用量を 375/125 mg 群及び 750/250 mg 群の 2 群から,
375/125 mg 群のみに変更した.Phase 2b Part の結果を用いた薬物動態/薬力学解析から,両投与
量による曝露間で本剤の有効性に明確な差はなく,375/125 mg 群で十分な抗ウイルス効果が確
認できたことから [39],本剤の申請用法・用量として低用量である 375/125 mg を選択すること
は妥当と判断した.高用量である 750/250 mg 群については,安全性解析及び副次的な位置付け
としての有効性解析を実施することとした.
なお,本剤の申請用法用量を 375/125 mg として,2022 年 2 月 25 日に製造承認販売申請を行
い,同年 7 月 20 日の薬事分科会・医薬品第二部会 (合同開催) において,Phase 3 Part 等の結果
をもって継続審議と判断されたが,承認される場合,用法用量を 375/125 mg とすることは妥当
と判断されている [39].
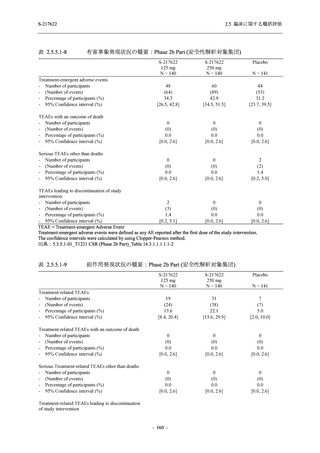
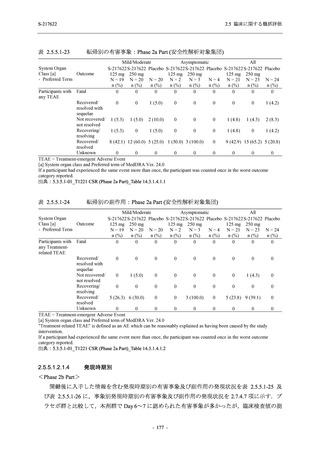
2. 主要解析の解析対象集団
主要評価項目及び主要な副次評価項目を対象とした主要解析における多重性調整の固定順序
法において,まず ITT 集団のうち,COVID-19 発症から無作為割付までの時間が 72 時間未満の
被験者に限定した解析対象集団で 375/125 mg 群とプラセボ群の比較を目的とした検定を行うこ
ととした.ここで有意な差が認められた場合には,引き続き,COVID-19 発症から無作為割付ま
での時間が 120 時間以内の被験者を対象とした ITT 集団全体を対象として検定を行うこととし
た.
- 128 -