











よむ、つかう、まなぶ。
資料2 事務局 提出資料 (10 ページ)
出典
| 公開元URL | https://www8.cao.go.jp/kisei-kaikaku/kisei/meeting/wg/2501_02medical/250428/medical04_agenda.html |
| 出典情報 | 規制改革推進会議 健康・医療・介護ワーキング・グループ(第4回 4/28)《内閣府》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
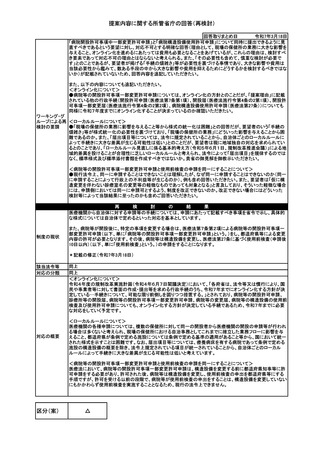
提案内容に関する所管省庁の回答
健康・医療・介護 WG関連
受付日
提案事項
具体的内容
別添
番号:5
所管省庁への検討要請日
令和5年11月17日 回答取りまとめ日
令和7年3月18日
No.64 低リスク遺伝子治療用製品の開発における治験開始前の環境影響評価の免除
欧州等の他のカルタヘナ議定書締約国と協調し、わが国においても少なくとも医療機器総合機構(PMDA)が公開
している生物多様性影響評価書(AAV記載例 令和5年2月版)に事例としてあるAAVベクターのように、承認済み
の複数の遺伝子治療用製品等医薬品で安全性が確認されている非増殖性で低病原性のリスクの低いベクター
については、治験開始前の評価を免除し、例えば米国と同様に承認申請時に審査を行うこととすべきである。
遺伝子治療用製品の開発においては、「遺伝子組換え生物等の使用等の規制による生物の多様性の確保に関
する法律(カルタヘナ法)」に基づき、治験開始前に環境影響評価を実施し、主務大臣(厚生労働大臣及び環境大
臣)の承認を得ることが求められている。現在、日本では治験開始前の申請書の提出から承認を得るまでの総事
務処理期間の中央値が5.0か月(2022年度参考値)である。一方、米国においては原則、治験開始前の申請は不
要とされ、承認審査の段階で環境影響評価の情報を添付し同時に審査を受けることとなっている。
このように、日本では米国に比べ治験開始に長い対応時間が必要となる。加えて、申請のための日本独自の資
料作成・手続も必要であり、これらの要因が、米国と国内の遺伝子治療薬開発品目数の大きな差の一因となって
いると考えられる。
提案理由
カルタヘナ法は生物の多様性を確保するための国際ルールである「カルタヘナ議定書」を適切に運用するために
定められた法律であるが、米国では議定書に批准していない。日本と同様、議定書に批准している欧州では、治
験開始前の環境影響評価が求められており、業界団体であるARM(Alliance for Regenerative Medicine)、EFPIA
(European Federation of Pharmaceutical Industries and Associations)、EuropaBio(European Association for
Bioindustries)から欧州委員会に対し、治験開始前の環境影響評価を免除すべきとの共同声明が公表されてい
る。
遺伝子治療用製品に使用されるアデノ随伴ウイルス(AAV)ベクターは、自己増殖能がなくヒトに対する病原性の
ないウイルスに由来し、非分裂細胞では遺伝子発現が長時間持続することから、遺伝子治療用ベクターとして有
力なベクターである。これまで、AAVベクターを利用した遺伝子治療用製品が複数承認されており、現在も様々な
治験が実施されているが、これまでAAVベクターを用いた遺伝子治療による環境リスクは認められていない。
近年、PMDAの取り組みにより、カルタヘナ法関連承認審査の運用改善がなされ、事務処理期間の大幅な短縮が
実現した。しかし、治験開始前の環境影響評価を要する欧州や日本よりも米国で遺伝子治療の開発が進んでお
り、国内での承認が遅れる、または承認されない、いわゆるドラッグ・ラグやドラッグ・ロスに繋がることも懸念され
る。
(要望実現により)ドラッグ・ラグやドラッグ・ロスの解消だけでなく、遺伝子治療用製品の開発における国際競争力
の強化に資することが期待できる。
提案主体
一般社団法人日本経済団体連合会
健康・医療・介護 WG関連
受付日
提案事項
具体的内容
別添
番号:5
所管省庁への検討要請日
令和5年11月17日 回答取りまとめ日
令和7年3月18日
No.64 低リスク遺伝子治療用製品の開発における治験開始前の環境影響評価の免除
欧州等の他のカルタヘナ議定書締約国と協調し、わが国においても少なくとも医療機器総合機構(PMDA)が公開
している生物多様性影響評価書(AAV記載例 令和5年2月版)に事例としてあるAAVベクターのように、承認済み
の複数の遺伝子治療用製品等医薬品で安全性が確認されている非増殖性で低病原性のリスクの低いベクター
については、治験開始前の評価を免除し、例えば米国と同様に承認申請時に審査を行うこととすべきである。
遺伝子治療用製品の開発においては、「遺伝子組換え生物等の使用等の規制による生物の多様性の確保に関
する法律(カルタヘナ法)」に基づき、治験開始前に環境影響評価を実施し、主務大臣(厚生労働大臣及び環境大
臣)の承認を得ることが求められている。現在、日本では治験開始前の申請書の提出から承認を得るまでの総事
務処理期間の中央値が5.0か月(2022年度参考値)である。一方、米国においては原則、治験開始前の申請は不
要とされ、承認審査の段階で環境影響評価の情報を添付し同時に審査を受けることとなっている。
このように、日本では米国に比べ治験開始に長い対応時間が必要となる。加えて、申請のための日本独自の資
料作成・手続も必要であり、これらの要因が、米国と国内の遺伝子治療薬開発品目数の大きな差の一因となって
いると考えられる。
提案理由
カルタヘナ法は生物の多様性を確保するための国際ルールである「カルタヘナ議定書」を適切に運用するために
定められた法律であるが、米国では議定書に批准していない。日本と同様、議定書に批准している欧州では、治
験開始前の環境影響評価が求められており、業界団体であるARM(Alliance for Regenerative Medicine)、EFPIA
(European Federation of Pharmaceutical Industries and Associations)、EuropaBio(European Association for
Bioindustries)から欧州委員会に対し、治験開始前の環境影響評価を免除すべきとの共同声明が公表されてい
る。
遺伝子治療用製品に使用されるアデノ随伴ウイルス(AAV)ベクターは、自己増殖能がなくヒトに対する病原性の
ないウイルスに由来し、非分裂細胞では遺伝子発現が長時間持続することから、遺伝子治療用ベクターとして有
力なベクターである。これまで、AAVベクターを利用した遺伝子治療用製品が複数承認されており、現在も様々な
治験が実施されているが、これまでAAVベクターを用いた遺伝子治療による環境リスクは認められていない。
近年、PMDAの取り組みにより、カルタヘナ法関連承認審査の運用改善がなされ、事務処理期間の大幅な短縮が
実現した。しかし、治験開始前の環境影響評価を要する欧州や日本よりも米国で遺伝子治療の開発が進んでお
り、国内での承認が遅れる、または承認されない、いわゆるドラッグ・ラグやドラッグ・ロスに繋がることも懸念され
る。
(要望実現により)ドラッグ・ラグやドラッグ・ロスの解消だけでなく、遺伝子治療用製品の開発における国際競争力
の強化に資することが期待できる。
提案主体
一般社団法人日本経済団体連合会