





































よむ、つかう、まなぶ。
資料No.2~2-1_日本薬局方の参考情報の改正(案)について (25 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/shingi2/0000174942_00008.html |
| 出典情報 | 薬事・食品衛生審議会 日本薬局方部会(令和5年度第1回 1/22)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
参考情報
13 .
1
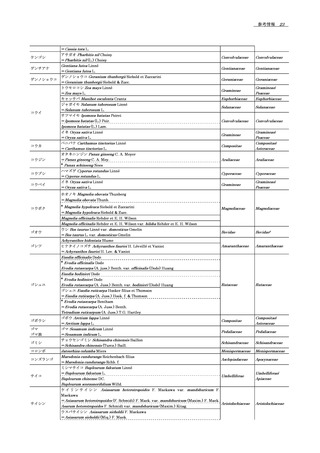
質に応じて経験的に選択される.シリカ,ポリマー又はハイブ
55
ることが多い.pHを下げ,それによりペプチドのプロトン化
2
リッド担体を基にした種々の孔径(8 ~ 100 nm)又は無細孔の
56
を促進する目的で,一般にギ酸や酢酸が移動相に添加される.
3
カラムは,十分な分離を与えることが示されてきた.粒子径が
57
緩衝液や塩は,シグナルを減少させることに加え,不揮発性の
4
2 μm未満のカラムが利用でき,一般的に3 ~ 5 μmの粒子径の
58
塩がイオン源に付着するため,使用は最小限にすべきである.
5
カラムよりも分離効率がよい.一般に,オクチル又はオクタデ
59
前述のように,TFAは,マトリックス干渉の一種であるイオ
6
シルシリル基を結合させた固定相がペプチドには最適である.
60
ンサプレッションを引き起こし,特にESIを用いた場合にペプ
7
30 nm又はそれより小さな細孔を持つオクタデシルシラン
61
チドのシグナルを抑制する可能性があるため,避けるべきであ
8
(C18)がペプチドマップの分離ステップで最もよく利用される
62
る.また,イオンサプレッションは糖ペプチドのイオン化効率
9
結合相である.
63
を抑制し,感度を低下させる.したがって,UVとMSの両方
10
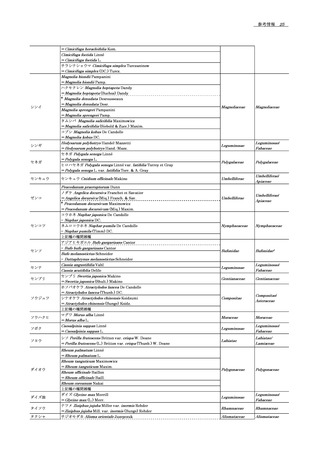
ペプチドのRP-HPLC分離に最も一般的な移動相は,有機溶
64
において最適な結果を得るためには,条件を最適化することが
11
媒としてアセトニトリルを含む水である.しかし,メタノール,
65
重要である.
12
2-プロパノール,又は1-プロパノールなどの他の有機溶媒
66
7.
13
も用いることができる.移動相にプロパノールなどの溶媒を用
67
ペプチドマップ法は相対比較の手法である.試料タンパク質
14
いることは,疎水性の高いペプチドを多く含む試料の分離に有
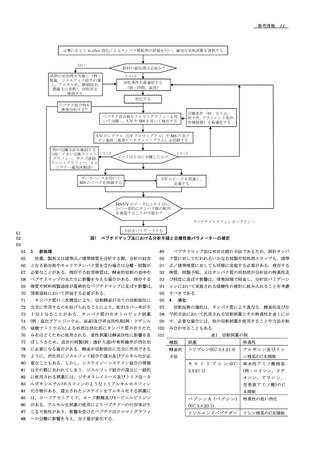
68
が意図するタンパク質であるかを確認するために,試料タンパ
15
用である.しかし,親水性又は短いペプチドはカラムのボイド
69
ク質のペプチドマップを標準品/標準物質を同様な前処理,分
16
容量を示す時間に溶出する可能性があることに留意する.酸,
70
離及び検出方法を用いて得られたペプチドマップと比較しなけ
17
塩基,緩衝塩及びイオンペア試薬のような移動相の添加剤は,
71
ればならない.保持時間,ピークレスポンス(ピーク面積又は
18
一般に,ペプチドの良好なクロマトグラフィー分離のために必
72
ピーク高さ),ピーク数及び全体的な溶出パターンの視覚的な
19
要である.最も一般的な移動相の添加剤はトリフルオロ酢酸
73
比較は,手順の最初のステップである.重要なピークのピーク
20
(TFA)であり,一般的には0.05 ~ 0.2%の濃度で用いられる.
74
レスポンス比及びピークの保持時間について,更に客観的解析
21
添加剤としてリン酸の使用はあまり一般的ではないが,紫外
75
を行うことが最良の方法である.もし試料タンパク質消化物及
22
(UV)検出器を用いる場合に有用である.揮発性の酸や塩は,
76
び標準品/標準物質の消化物の全ての重要なピークが同じ保持
23
質量分析計による検出との親和性を改善するために移動相に用
77
時間及びピークレスポンス比を示したなら,試料タンパク質の
24
いることができる.TFAはペプチドの分離の質に非常に良い
78
同一性が確認される.例えば,モノクローナル抗体試料は,共
25
影響を及ぼすが,質量分析計による検出の感度は,イオンサプ
79
通のFcペプチドを含んでおり,ペプチドマップ試験の際には
26
レッション効果により悪影響を受ける.ギ酸,酢酸又はこれら
80
参照ピークとして用いられている.参照ペプチドを試料消化物
27
をTFAと共に用いると,イオンサプレッションを抑制するこ
81
に添加し,重要なピークのピークレスポンス比と保持時間をあ
28
とにより質量分析計の感度を向上することができる.クロマト
82
らかじめ設定された判定基準と比較することが可能である.選
29
グラフィーカラムの温度調節は,良好な再現性を得るために必
83
択される比較方法は,得られるペプチドマップの複雑さと個々
30
要である.逆相カラムにおいて分離は一般に温度の上昇と共に
84
の確認試験の目的(例:同一施設で製造される別のタンパク質
31
向上するため,カラム温度は,ペプチド分離の最適化やある種
85
医薬品との区別や同じタンパク質医薬品の変異体との区別)に
32
のペプチドの保持や溶出を改善するために用いられることがあ
86
おいて求められる特異性によって異なる.
33
る.
87
高い特異性が求められる場合,質量分析を日常的な分析にお
34
6.
88
いて用いることで,ペプチドの修飾,切断,切断ミス,不純物
検出
データ解析
35
RP-HPLCは,確認試験としてのペプチドマップ法で用いら
89
及び分離されずに一つのピークとして共溶出したピークに関す
36
れる最も一般的な分離方法であり,最も一般的な検出方法は,
90
る知見を得ることができる.
37
214 nmでのUV光吸収である.タンパク質の消化により生じた
91
8.
38
ペプチドは,より長波長(例:280 nm)の光を吸収する芳香族
92
ペプチドマップ法の手順の開発の間に,システム適合性の基
39
側鎖を持つアミノ酸を含まない場合があるので,タンパク質の
93
準及び分析法バリデーションの判定基準の選択につながる知識
バリデーション実施前の留意事項
40
配列カバー率を確保するには,移動相によるバックグラウンド
94
や経験が得られる.バリデーション実施前の最終レビューによ
41
を最小化するように注意し,214 nm(ペプチド結合が吸収する
95
り,手順がバリデーションの準備ができていることを確認し,
42
光の波長)での検出が不可欠である.また,その他の検出方法
96
基準を満たさないリスクを減らすことができる.一般的な手順
43
も適切である.
97
として,ペプチドマップ法は,広範囲な試験デザイン,試験目
44
UV検出の限界は,ペプチドの構造に関する情報が得られな
98
的及び性能に関する要求を含んでいる.したがって,一般的な
45
いことである.質量分析は,ペプチドが同時に溶出した場合の
99
文書にて,特定のシステム適合性やバリデーション基準を規定
46
選択性に加えて,ペプチドの同定に役立つ質量情報を提供する
100
することは不可能である.バリデーション開始前に次の要素に
47
有用な検出方法である.ほとんどの分析目的において,RP-
101
ついて評価することが推奨される.
48
HPLCからの溶出液は,移動相が質量分析計に適している場合
102
ペプチドマップ法の日常的な測定における質量分析の利用は
49
には,直接質量分析計に導入することができる.移動相に特有
103
本参考情報には記載していないが,ペプチドマップ法の開発段
50
の留意事項は,選択したイオン化方法による.エレクトロスプ
104
階におけるペプチドの構造同定に質量分析を適用することは最
51
レーイオン化法(ESI)は,タンパク質やペプチドを質量分析計
105
良の方法である.質量分析による検出は,性能に関する以下の
52
に導入する最も一般的な方法であり,揮発性の水溶媒混合液を
106
パラメーターを評価するために利用される.
53
用いた際に最もよいイオン化効率が得られる.ESI-MSを用い
107
8.1.
54
たペプチドマップ法では,ポジティブイオンモードが用いられ
108
配列カバー率
配列カバー率は,目的のタンパク質配列について,ペプチド
13 .
1
質に応じて経験的に選択される.シリカ,ポリマー又はハイブ
55
ることが多い.pHを下げ,それによりペプチドのプロトン化
2
リッド担体を基にした種々の孔径(8 ~ 100 nm)又は無細孔の
56
を促進する目的で,一般にギ酸や酢酸が移動相に添加される.
3
カラムは,十分な分離を与えることが示されてきた.粒子径が
57
緩衝液や塩は,シグナルを減少させることに加え,不揮発性の
4
2 μm未満のカラムが利用でき,一般的に3 ~ 5 μmの粒子径の
58
塩がイオン源に付着するため,使用は最小限にすべきである.
5
カラムよりも分離効率がよい.一般に,オクチル又はオクタデ
59
前述のように,TFAは,マトリックス干渉の一種であるイオ
6
シルシリル基を結合させた固定相がペプチドには最適である.
60
ンサプレッションを引き起こし,特にESIを用いた場合にペプ
7
30 nm又はそれより小さな細孔を持つオクタデシルシラン
61
チドのシグナルを抑制する可能性があるため,避けるべきであ
8
(C18)がペプチドマップの分離ステップで最もよく利用される
62
る.また,イオンサプレッションは糖ペプチドのイオン化効率
9
結合相である.
63
を抑制し,感度を低下させる.したがって,UVとMSの両方
10
ペプチドのRP-HPLC分離に最も一般的な移動相は,有機溶
64
において最適な結果を得るためには,条件を最適化することが
11
媒としてアセトニトリルを含む水である.しかし,メタノール,
65
重要である.
12
2-プロパノール,又は1-プロパノールなどの他の有機溶媒
66
7.
13
も用いることができる.移動相にプロパノールなどの溶媒を用
67
ペプチドマップ法は相対比較の手法である.試料タンパク質
14
いることは,疎水性の高いペプチドを多く含む試料の分離に有
68
が意図するタンパク質であるかを確認するために,試料タンパ
15
用である.しかし,親水性又は短いペプチドはカラムのボイド
69
ク質のペプチドマップを標準品/標準物質を同様な前処理,分
16
容量を示す時間に溶出する可能性があることに留意する.酸,
70
離及び検出方法を用いて得られたペプチドマップと比較しなけ
17
塩基,緩衝塩及びイオンペア試薬のような移動相の添加剤は,
71
ればならない.保持時間,ピークレスポンス(ピーク面積又は
18
一般に,ペプチドの良好なクロマトグラフィー分離のために必
72
ピーク高さ),ピーク数及び全体的な溶出パターンの視覚的な
19
要である.最も一般的な移動相の添加剤はトリフルオロ酢酸
73
比較は,手順の最初のステップである.重要なピークのピーク
20
(TFA)であり,一般的には0.05 ~ 0.2%の濃度で用いられる.
74
レスポンス比及びピークの保持時間について,更に客観的解析
21
添加剤としてリン酸の使用はあまり一般的ではないが,紫外
75
を行うことが最良の方法である.もし試料タンパク質消化物及
22
(UV)検出器を用いる場合に有用である.揮発性の酸や塩は,
76
び標準品/標準物質の消化物の全ての重要なピークが同じ保持
23
質量分析計による検出との親和性を改善するために移動相に用
77
時間及びピークレスポンス比を示したなら,試料タンパク質の
24
いることができる.TFAはペプチドの分離の質に非常に良い
78
同一性が確認される.例えば,モノクローナル抗体試料は,共
25
影響を及ぼすが,質量分析計による検出の感度は,イオンサプ
79
通のFcペプチドを含んでおり,ペプチドマップ試験の際には
26
レッション効果により悪影響を受ける.ギ酸,酢酸又はこれら
80
参照ピークとして用いられている.参照ペプチドを試料消化物
27
をTFAと共に用いると,イオンサプレッションを抑制するこ
81
に添加し,重要なピークのピークレスポンス比と保持時間をあ
28
とにより質量分析計の感度を向上することができる.クロマト
82
らかじめ設定された判定基準と比較することが可能である.選
29
グラフィーカラムの温度調節は,良好な再現性を得るために必
83
択される比較方法は,得られるペプチドマップの複雑さと個々
30
要である.逆相カラムにおいて分離は一般に温度の上昇と共に
84
の確認試験の目的(例:同一施設で製造される別のタンパク質
31
向上するため,カラム温度は,ペプチド分離の最適化やある種
85
医薬品との区別や同じタンパク質医薬品の変異体との区別)に
32
のペプチドの保持や溶出を改善するために用いられることがあ
86
おいて求められる特異性によって異なる.
33
る.
87
高い特異性が求められる場合,質量分析を日常的な分析にお
34
6.
88
いて用いることで,ペプチドの修飾,切断,切断ミス,不純物
検出
データ解析
35
RP-HPLCは,確認試験としてのペプチドマップ法で用いら
89
及び分離されずに一つのピークとして共溶出したピークに関す
36
れる最も一般的な分離方法であり,最も一般的な検出方法は,
90
る知見を得ることができる.
37
214 nmでのUV光吸収である.タンパク質の消化により生じた
91
8.
38
ペプチドは,より長波長(例:280 nm)の光を吸収する芳香族
92
ペプチドマップ法の手順の開発の間に,システム適合性の基
39
側鎖を持つアミノ酸を含まない場合があるので,タンパク質の
93
準及び分析法バリデーションの判定基準の選択につながる知識
バリデーション実施前の留意事項
40
配列カバー率を確保するには,移動相によるバックグラウンド
94
や経験が得られる.バリデーション実施前の最終レビューによ
41
を最小化するように注意し,214 nm(ペプチド結合が吸収する
95
り,手順がバリデーションの準備ができていることを確認し,
42
光の波長)での検出が不可欠である.また,その他の検出方法
96
基準を満たさないリスクを減らすことができる.一般的な手順
43
も適切である.
97
として,ペプチドマップ法は,広範囲な試験デザイン,試験目
44
UV検出の限界は,ペプチドの構造に関する情報が得られな
98
的及び性能に関する要求を含んでいる.したがって,一般的な
45
いことである.質量分析は,ペプチドが同時に溶出した場合の
99
文書にて,特定のシステム適合性やバリデーション基準を規定
46
選択性に加えて,ペプチドの同定に役立つ質量情報を提供する
100
することは不可能である.バリデーション開始前に次の要素に
47
有用な検出方法である.ほとんどの分析目的において,RP-
101
ついて評価することが推奨される.
48
HPLCからの溶出液は,移動相が質量分析計に適している場合
102
ペプチドマップ法の日常的な測定における質量分析の利用は
49
には,直接質量分析計に導入することができる.移動相に特有
103
本参考情報には記載していないが,ペプチドマップ法の開発段
50
の留意事項は,選択したイオン化方法による.エレクトロスプ
104
階におけるペプチドの構造同定に質量分析を適用することは最
51
レーイオン化法(ESI)は,タンパク質やペプチドを質量分析計
105
良の方法である.質量分析による検出は,性能に関する以下の
52
に導入する最も一般的な方法であり,揮発性の水溶媒混合液を
106
パラメーターを評価するために利用される.
53
用いた際に最もよいイオン化効率が得られる.ESI-MSを用い
107
8.1.
54
たペプチドマップ法では,ポジティブイオンモードが用いられ
108
配列カバー率
配列カバー率は,目的のタンパク質配列について,ペプチド