









よむ、つかう、まなぶ。
【資料2】柏谷構成員提出資料 (7 ページ)
出典
| 公開元URL | https://www.mhlw.go.jp/stf/newpage_35743.html |
| 出典情報 | 創薬力の強化・安定供給の確保等のための薬事規制のあり方に関する検討会(第4回 10/13)《厚生労働省》 |
ページ画像
ダウンロードした画像を利用する際は「出典情報」を明記してください。
低解像度画像をダウンロード
プレーンテキスト
資料テキストはコンピュータによる自動処理で生成されており、完全に資料と一致しない場合があります。
テキストをコピーしてご利用いただく際は資料と付け合わせてご確認ください。
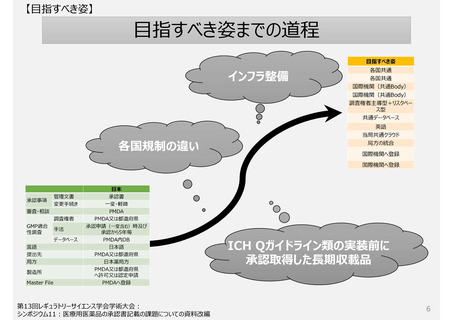
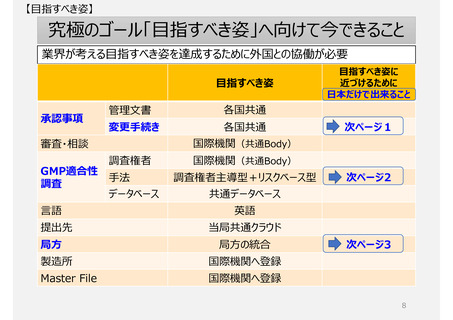
【日本の現状:課題】
課
題
日本の独自制度の実態
生じている問題
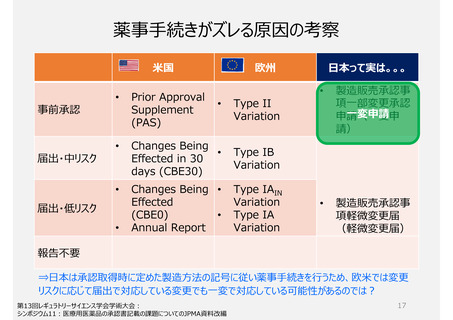
1. 製造方法(規格及び試験方法等含む)を変更する手続き制度
一部変更承認申請(一変申請)が行われる場合の変更後製
品への切替えタイミングが承認日(又は6カ月以内の企業が希望した
日)のみ
品質関係の変更は様々あるが、薬事手続きの区分は2区分
切替日に向けた生産調整の難易度
の高さ(欧米は新旧並行出荷可能)。
同じ変更でも日本は、欧米より時間
(特にバイオ医薬品)と手間がかか
る(出荷時期の予見性低下)。
変更手続き区分の間違いによる品
質事案の発生が防止できない。(欧
(一変申請/軽微変更届出)
軽微変更届出後、内容の妥当性については、後に行われる
一変審査において確認される。
承認事項と変更手続き区分の特定の仕方
日本は承認書に記載された文章一言一句すべて、
かつ、変更手続きの区分を判別する指標は、文章や各パラ
メーターにつけられた変更の区分を示す記号(承認時に特定)
米は提出直後に確認)
欧米の薬事手続きと異なる場合が
ある(欧米は、変更時点のリスク評価によってリ
スクレベルを判断、保管のみの製造所の取扱
い)。
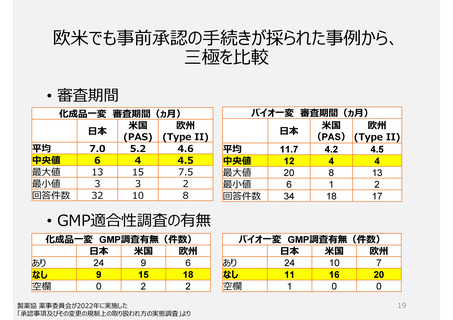
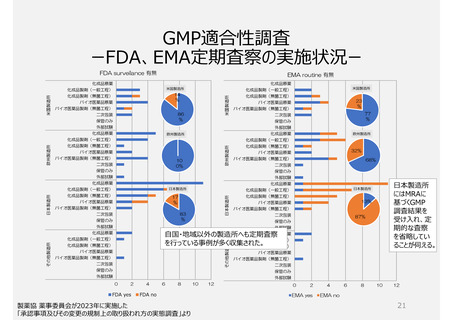
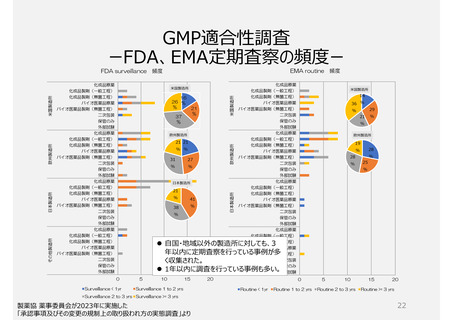
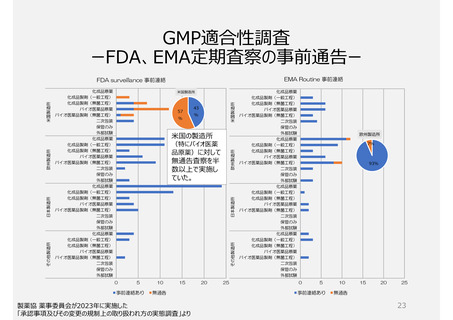
2. GMP適合性調査制度
承認前の調査は、承認申請品目に関係する全ての製造所に
ついて調査が行われ、承認後は5年毎に調査を受けなければ
ならない。
いずれも企業が調査申請をする制度
制度上、低リスクの製造所でも調査
頻度が高い場合がある(GMP調査経験
や製造所のリスクを考慮していないため)。
PMDAによる実地調査率が低い。
3.欧米薬局方(USP、Ph.Eur.)収載品の取扱い
他国の局方収載品は、試験の手順を詳細に書き下す形式
日本独自の局方に基づいた原料調達が必要
日本で個別に審査・承認を経ないと
製品出荷できない。
独自原料調達によるコスト増・納期
7
遅延
課
題
日本の独自制度の実態
生じている問題
1. 製造方法(規格及び試験方法等含む)を変更する手続き制度
一部変更承認申請(一変申請)が行われる場合の変更後製
品への切替えタイミングが承認日(又は6カ月以内の企業が希望した
日)のみ
品質関係の変更は様々あるが、薬事手続きの区分は2区分
切替日に向けた生産調整の難易度
の高さ(欧米は新旧並行出荷可能)。
同じ変更でも日本は、欧米より時間
(特にバイオ医薬品)と手間がかか
る(出荷時期の予見性低下)。
変更手続き区分の間違いによる品
質事案の発生が防止できない。(欧
(一変申請/軽微変更届出)
軽微変更届出後、内容の妥当性については、後に行われる
一変審査において確認される。
承認事項と変更手続き区分の特定の仕方
日本は承認書に記載された文章一言一句すべて、
かつ、変更手続きの区分を判別する指標は、文章や各パラ
メーターにつけられた変更の区分を示す記号(承認時に特定)
米は提出直後に確認)
欧米の薬事手続きと異なる場合が
ある(欧米は、変更時点のリスク評価によってリ
スクレベルを判断、保管のみの製造所の取扱
い)。
2. GMP適合性調査制度
承認前の調査は、承認申請品目に関係する全ての製造所に
ついて調査が行われ、承認後は5年毎に調査を受けなければ
ならない。
いずれも企業が調査申請をする制度
制度上、低リスクの製造所でも調査
頻度が高い場合がある(GMP調査経験
や製造所のリスクを考慮していないため)。
PMDAによる実地調査率が低い。
3.欧米薬局方(USP、Ph.Eur.)収載品の取扱い
他国の局方収載品は、試験の手順を詳細に書き下す形式
日本独自の局方に基づいた原料調達が必要
日本で個別に審査・承認を経ないと
製品出荷できない。
独自原料調達によるコスト増・納期
7
遅延